
基于M iRf ilter系统的毛果杨 m iRNA预测
2010-10-16 07:23:20赵洁苑龚云路王翼飞
上海大学学报(自然科学版) 2010年4期
赵洁苑, 龚云路, 王翼飞
(上海大学 理学院,上海 200444)
基于M iRf ilter系统的毛果杨 m iRNA预测
赵洁苑, 龚云路, 王翼飞
(上海大学 理学院,上海 200444)
从参数训练、参数范围训练、候选成熟体打分等方面改进 miRNA预测系统M iRfilter,使其适应拥有更长前体的植物miRNA的预测.预测毛果杨基因组上的 miRNA,并对系统进行精度检验.利用M iRfilter系统共预测出 3 860条候选 miRNA;在 110个正样本中,正确识别 91条前体和 80条成熟体,前体预测精度为 82.73%,成熟体预测精度为 72.73%;在毛果杨第 4号染色体 (LG_Ⅳ)上得到的 1 968个负样本中,有 12个数据可认为是 miRNA,假阳性率为0.61%.
植物 miRNA;M iRfilter;毛果杨基因组;一类分类法;K-最近邻分类器 (KNN)
Abstract:Thispaper improves theMiRfilter system by parameter,range of parameter and score in mature miRNAs in order to predict miRNAs in plants.We predict miRNAs in Populus trichocarpa genome and use itspositive and negative samples to examine the accuracy of MiRfilter’sp rediction.M iRfilter predicts 3 860 Populus trichocarpa miRNA candidates in all.It correctly identifies91 p recursors and 80 matures in 110 positive samp les.Accuracies of the precursor and mature prediction reach 82.73%and 72.73%,respectively.We find 12 false positive miRNA s from 1 968 negative samp les in LG_Ⅳchromosome,and the false positive rate reaches0.61%.
Key words:plant miRNA;MiRfilter;Populus trichocarpa genome;one-class methods;K-nearest neighbor(KNN)
miRNA是一类长度约为 20~24 nt(少数小于20 nt)的内源性非编码调控单链小分子 RNA,由一段具有发夹结构的单链 RNA前体 (pre-miRNA)剪切后生成.成熟的 miRNA 5′端为单磷酸基,3′端为羟基,通过与其靶 mRNA分子的 3′端非编码区域 (3′-untranslated region,3′UTR)互补匹配来抑制该mRNA分子的翻译.miRNA基因以单拷贝、多拷贝或基因簇等多种形式存在于基因组中,而且绝大部分定位于基因间隔区.miRNA在生物体中的基因表达具有进化保守性、时序性和组织特异性等特点,显示了其在控制个体发育、决定细胞命运和分化中的特定功能[1].到 2009年 2月为止,miRBase(http:∥microrna.sanger.ac.uk/cgi-bin/sequences/)数据库已发布了 8 619种 miRNA.
除一些基本特征外,植物 miRNA和动物miRNA有明显区别[2-5].植物 miRNA前体比动物的更长、更复杂,通常为数百核苷酸;不同于动物 miRNA的加工来自于蛋白质编码基因内含子,大部分植物miRNA前体产生于其自身的转录单元;植物 miRNA以单基因形式为多,基因簇内的 miRNA排列也相对较松散;植物 miRNA的靶序列还包括蛋白质编码区[2].研究植物 miRNA的功能可以通过以下两种方法:①导入抗miRNA的靶基因或上调 miRNA的表达,分析植物出现的表型变化;②用生化与分子生物学方法,如 cDNA末端快速扩增 (rapid amplification of cDNA ends,RACE)技术,测定 miRNA指导下靶mRNA剪切反应的精确位点[2].研究表明,植物miRNA主要通过以下 3种途径来调节基因的表达:①通过碱基间互补配对直接结合于靶基因 mRNA上,从而导致靶基因的特异性剪切;②miRNA介导的翻译阻遏;③miRNA介导的翻译沉默[4].由此可见,植物miRNA的正常表达是植物正常生长发育所必需的,它所调节的靶基因控制着植物生长发育的各个方面,包括根、叶、花等形态发生、细胞分化、疏导组织形成等,也在调节植物对环境胁迫如干旱、盐害和养分胁迫反应等方面起着重要的作用[2-3].
miRNA可以通过 cDNA克隆测序的方法加以识别.一些实验室已经建立了不同组织、不同发育时期或不同生长条件下的 miRNA基因文库.然而,真核生物组织中有些 miRNA的丰度较低,其表达又具有时序性和组织特异性,使得克隆法分离 miRNA十分困难.通过计算预测miRNA成为miRNA发现的一个有效方法,该方法以基因组序列和计算机程序鉴定为基础进行科学预测和鉴定,弥补了 cDNA克隆测序方法中的不足.根据计算预测方法的本质,可分为以下 5种[6]:同源片段搜索方法、基于比较基因组学的预测方法[7-8]、基于序列和结构特征打分的预测方法[9]、结合作用靶标的预测方法、基于机器学习的预测方法[10-11].
MiRfilter系统是本实验室自主开发的一个用于预测miRNA的自动化软件,不依赖物种的同源性,仅根据物种已知 miRNA的固有信息进行预测,属于基于序列和结构特征打分的预测方法,在病毒miRNA识别中具有良好的预测精度[12].然而,由于植物 miRNA的序列结构特征较病毒来说更为多样和复杂,直接使用M iRfilter系统进行预测的效果并不理想.为了使MiRfilter系统能够适应植物 miRNA的预测,本研究从参数训练、参数范围训练、候选成熟体打分等方面对系统进行改进,并应用于毛果杨(Populus trichocarpa)的 miRNA预测.
1 材料与方法
1.1 M iRf ilter系统简介
M iRfilter系统的预测步骤层次分明,整个过程分为 4个阶段[12]:①在预测之前,对待测物种的基因组序列及已知 miRNA序列进行预处理;②根据预处理后的训练集界定训练参数和参数范围;③对预测区域作二级结构模拟,从中提取合格的发夹结构;④根据训练得到的参数范围,从合格的发夹结构中筛选候选miRNA成熟体序列和前体序列.系统具体流程如图1所示.

图1 M iRf ilter系统流程图Fig.1 Flow char t of the M iRf ilter system
1.2 M iRf ilter系统改进
1.2.1 参数训练
在前体参数中,由于植物前体长度跨度较大,预测出的前体与miRBase数据库给出的前体在序列两端可能存在一定的差异,该差异会同时影响最小自由能的数值,使依赖于前体长度和自由能这两个参数的MFEL[13]参数变化过大.用MFEL筛选前体,有时会将已知的 miRNA前体排除出去.因此,本研究去掉MFEL参数,另外增加 3个新参数,即定位前体(不包括发夹结构尾部的前体,如图2所示)的长度、茎区配对个数以及它们的比值,使筛选不受前体序列两端差异的影响,提高预测精度;还统计了两条茎上配对碱基的个数,以此替代原来用于判断合格发夹结构的标准——18 nt.
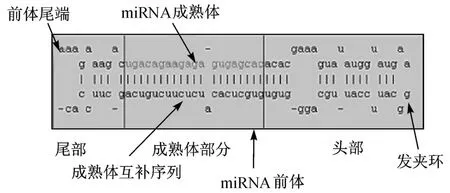
图2 m iRNA前体二级结构示意图(ptc-M IR156a发夹结构)Fig.2 Pre-m iRNA stem-loop(ptc-M IR 156a ha irp in structure)
在成熟体参数中,miRNA序列的首字母特征明显,倾向于以 U开始[2],因此,本研究添加成熟体序列首字母参数,作为参与成熟体打分的一个变量;补充成熟体所在臂参数、发夹环与成熟体之间的配对碱基个数、成熟体与前体尾端之间的距离,这些参数与原有的发夹环与成熟体之间的距离参数一起界定候选成熟体在前体上的位置.
另外,本研究增加了一组成熟体互补序列参数,包括互补序列的长度、最大内环大小、平均内环大小、内环个数.这些参数能更好地反映成熟体互补序列本身的序列结构特征,以及成熟体序列与其在结构上的对称性质.
最终,本研究确定以下三类描述 miRNA前体及成熟体特征的参数.
(1)前体参数:H1为前体序列长度 (Prelen);H2为发夹环大小 (Hplen);H3为发夹结构最小自由能(Energy);H4为前体茎区配对碱基个数 (paNum);H5为定位前体序列长度 (MarPrelen);H6为定位前体茎区配对碱基个数 (MarPaNum);H7为定位前体茎区配对碱基个数与其长度之比 (MarPAPL=MarPaNum/MarPrelen).
(2)成熟体参数:M1为成熟体序列长度(Marlen);M2为成熟体序列首字母 (Marst1);M3为成熟体序列中 C+G含量 (cgCon);M4为成熟体序列在发夹结构中的位置,即左臂或右臂 (Arm);M5为发夹环与成熟体之间的距离 (Dist1);M6为发夹环与成熟体之间的配对碱基个数(Dist1P);M7为成熟体与前体尾端之间的距离 (Dist2);M8为发夹结构茎区内成熟序列中不配对碱基的个数 (upNum);M9为发夹结构茎区内成熟序列的两端处不配对碱基的个数(TerUpNum);M10为发夹结构茎区内成熟序列中最大内环的大小(InlpMax);M11为发夹结构茎区内成熟序列中内环的平均大小 (InlpAvg);M12为发夹结构茎区内成熟序列中内环的个数(InlpNum).
(3)成熟体互补序列参数:P1为成熟体互补序列长度(Parlen);P2为发夹结构茎区内成熟体互补序列中最大内环的大小 (Par InlpMax);P3为发夹结构茎区内成熟体互补序列中内环的平均大小(Par InlpAvg);P4为发夹结构茎区内成熟体互补序列中内环的个数(Par InlpNum).
1.2.2 参数范围训练
在对 1.2.1节参数进行范围界定时,依然采用按总数据量的 3%删除最大或最小参数值的方法.根据每个参数的实际意义选择适合的删除原则,使得到的范围更具有针对性.具体可分为以下 4种情况:
(1)同时删除最大值和最小值,得到一个范围,适合一般参数,包括 H1,H2,H4,H5,H6,H7,M3,M7,P1.
(2)只删除最大值,得到一个范围,适合一般认为参数值越小越好的参数,包括 H3,M8,M9,M10,M11,M12,P2,P3,P4.
(3)取中位数,适合只需用一个均值描述整体的参数,包括M1(使用中位数是为了避免再次取整).
(4)将左臂和右臂作为两类分别统计,适合在这两类中范围相差较大的参数,避免其中一类范围扩大,包括 M5,M6.
表 1列出了毛果杨各参数的范围.
1.2.3 M iRNA预测
本研究根据修改后的新参数调整了预测前体和成熟体的筛选标准.在最后预测成熟体的过程中,一个候选前体上可能会有多个符合标准的成熟体被保留下来.为此,引入一个打分机制,为每一个候选前体上预测出的成熟体打分,从中挑选出得分最佳的成熟体作为该前体的候选成熟体.具体打分方法采用最近邻一类分类法.
一般而言,两类分类法 (two-classmethods)需要考虑正样本和负样本两组数据,通过一定的算法学习这两类样本,从而构建一个能够区分它们的分类器.使用两类分类法识别miRNA,是将已知的miRNA作为正样本的同时,还需要人为地构造一组非miRNA的负样本数据.但是负样本的选择具有一定的难度,如果选出的负样本并不适合,就会显著影响分类器的表现或者产生巨大误差.另一方面,一类分类法 (one-class methods)只需要考虑目标类 (正样本)的信息,就可以构建一个能够识别目标类样本并丢弃其他非目标类样本的分类器,避免了人为构造负样本数据[14].因此,在无法确定负样本的情况下,采用一类分类法识别新的miRNA.
最近邻一类分类法(one-class K-nearest neighbor classifier,OC-KNN)是一种修正了已知的最近邻两类分类法,使其只学习正样本数据的分类方法[14].该算法存储所有的训练样本(正样本)y,将其作为邻居集;对于一个给定的测试样本 z,计算 z到邻居集中所有邻居 y的距离 d(z,y);将 k个最近邻居距离的平均值作为 z的得分,当得分满足一定条件时,将z归为目标类.
在实际应用中,将已知的 miRNA作为训练样本y,将预测出的 miRNA作为测试样本 z,每个样本包含成熟体及其互补序列的序列参数和结构参数,即H7,M2,M3,M8,M9,M10,M11,M12,P1,P2,P3,P4共12个变量;取 k=1,保留满足以下打分公式的测试样本:
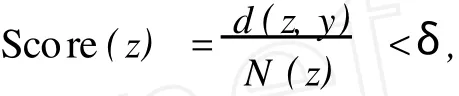
式中,d(z,y)采用欧拉距离,且变量在计算之前先标准化;N(z)为测试样本 z所在的候选前体上所有预测出的miRNA的个数;阈值δ可根据已知 miRNA的得分进行选取.在每个候选前体上选取得分最低的,即与已知 miRNA相似度最高的成熟体,将其作为该候选前体的候选成熟体.表 2列出了毛果杨各条染色体选取的δ值.

表 1 毛果杨参数范围Table 1 Ranges of Populus trichocarpa’s param eter s
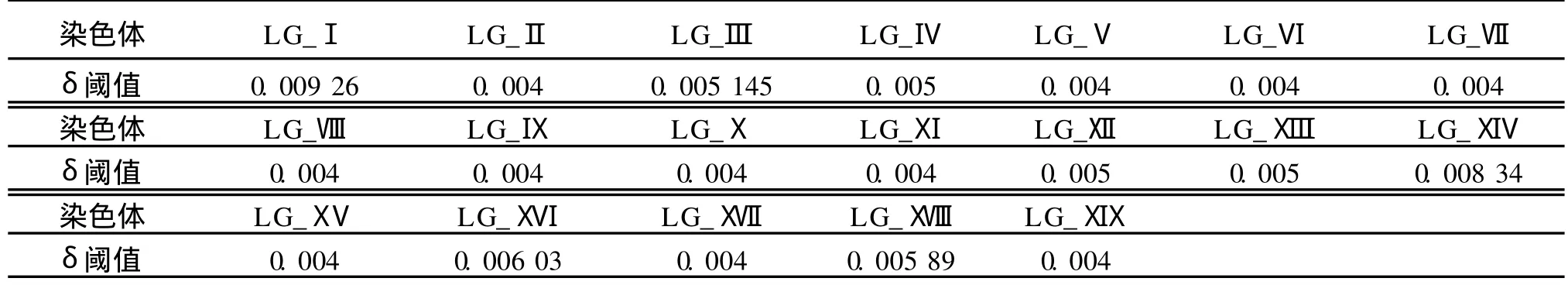
表 2 毛果杨各染色体δ值Table 2 δof Populus trichocarpa’s each chrom osom e
1.3 数据集
本研究使用改进的MiRfilter系统在毛果杨基因组序列中预测miRNA,并根据预测结果对该系统的预测精度进行检验.毛果杨基因组 19对染色体 4.8亿个碱基的测序工作已于 2004年 9月 21日完成,这是林木上第一个、植物上继拟南芥和水稻之后第三个进行基因组测序的物种[15],其基因组序列 (版本 1.1)及相关注释文件可从杨树基因网站 JGI(http://www.jgi.doe.gov/poplar/)上获得.毛果杨已知的 miRNA数据取自 miRBase数据库 (2008年 8月).本研究保留 19对染色体中前体二级结构只含有一个发夹环、成熟体长度为 21 nt的 miRNA序列,共 110条前体上的 110条成熟体,将其作为正样本数据;负样本数据选用在毛果杨第 4号染色体 (LG_Ⅳ)的外显子部分中预测出的 1 968条可能的成熟体.
1.4 评价标准
对于每一个测试样本,只可能属于以下 4种类型之一:正确识别的正样本 TP、正确识别的负样本TN、本来是负样本却被识别为正样本 (假阳性样本)FP、本来是正样本却被识别为负样本 (假阴性样本)FN.用 N表示样本总数,Q表示总预测精度,QP表示正样本的预测精度,QN表示负样本的预测精度,FPR表示假阳性预测率,FNR表示假阴性预测率,MCC表示Matthew相关系数,分别定义如下[12]:

2 结果与讨论
本研究在毛果杨非外显子序列中预测出 3 860条成熟体,对应 3 860条前体;在 110个正样本中正确识别出 91条已知前体和 80条已知成熟体,前体预测精度达 82.73%,成熟体预测精度达 72.73%,表 3为各条染色体的预测情况.在未被识别出的 30个miRNA中,有 16个 miRNA的前体已被预测出来,但由于存在得分更低的成熟体序列而被排除;有14个miRNA因没有预测出其前体而被排除,表 4列出了未被成功识别出的毛果杨 miRNA.根据毛果杨第 4号染色体的阈值δ,对 1 968个负样本数据进行筛选,最终有 12个数据被认为是 miRNA,假阳性率为 0.61%,具体假阳性数据见表 5.表 6为改进后的M iRfilter系统具体的预测精度.
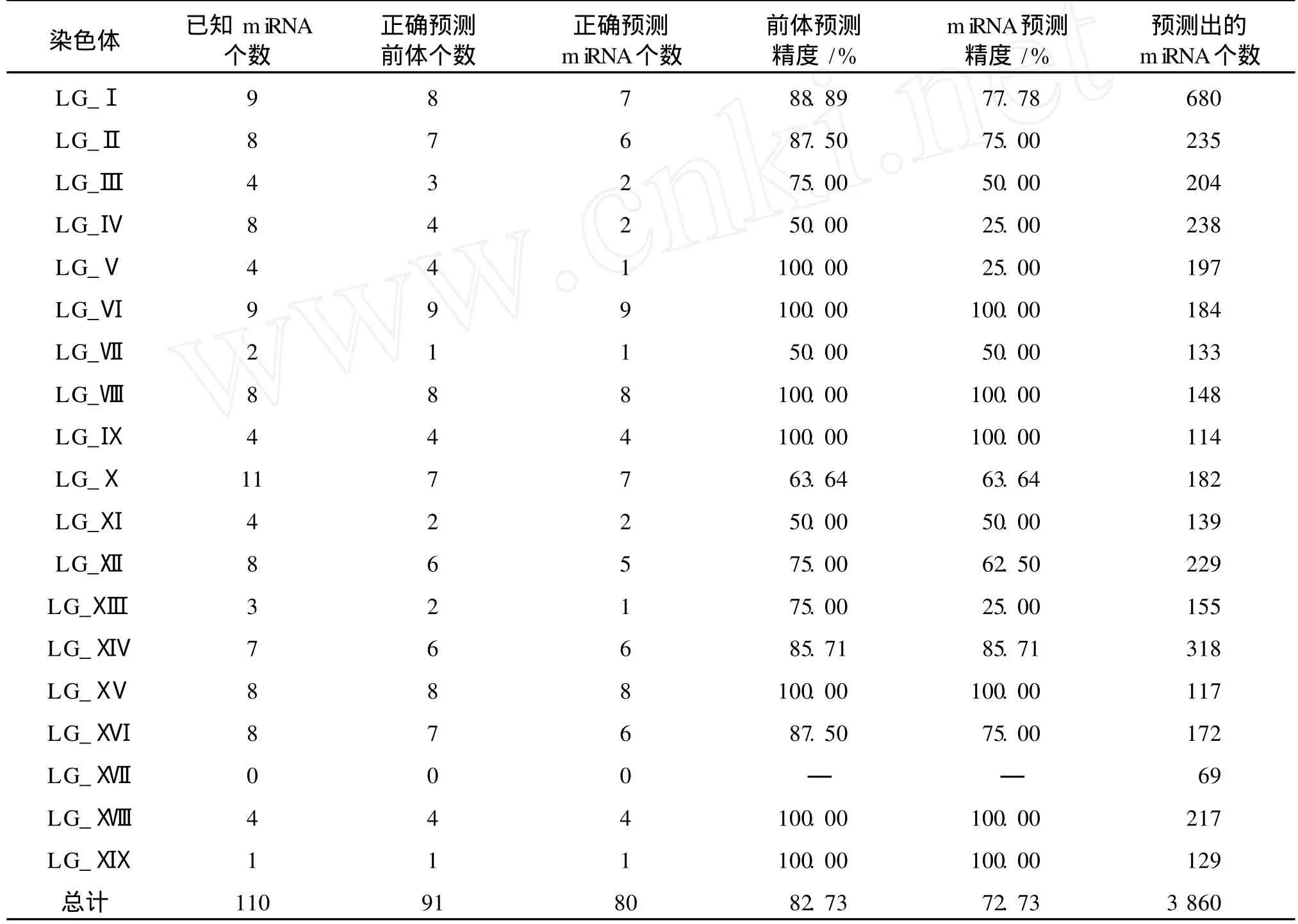
表 3 毛果杨各染色体的预测结果和预测精度Table 3 Pred iction resultsand accuracy of Populus trichocarpa’s each chromosome
序列分析发现,病毒 miRNA之间的序列相似性很低,很少存在同源序列.对于很多病毒而言,它们只存在进化距离很远的直系同源成员.类似的问题也发生在高等真核生物中.迄今为止,具有完整基因组序列且与拟南芥进化距离相对最近的物种是水稻,而水稻与拟南芥基因组早在 2亿年前就已经分化.具有完整基因组序列且与人类进化距离相对最近的物种是黑猩猩,而黑猩猩与人类的基因组也早在 4百万年前就已经分化[6].因此,不依赖序列保守性的 M iRfilter系统适用于各种生物的miRNA预测,它是发现非同源、物种特异 miRNA的有效途径.
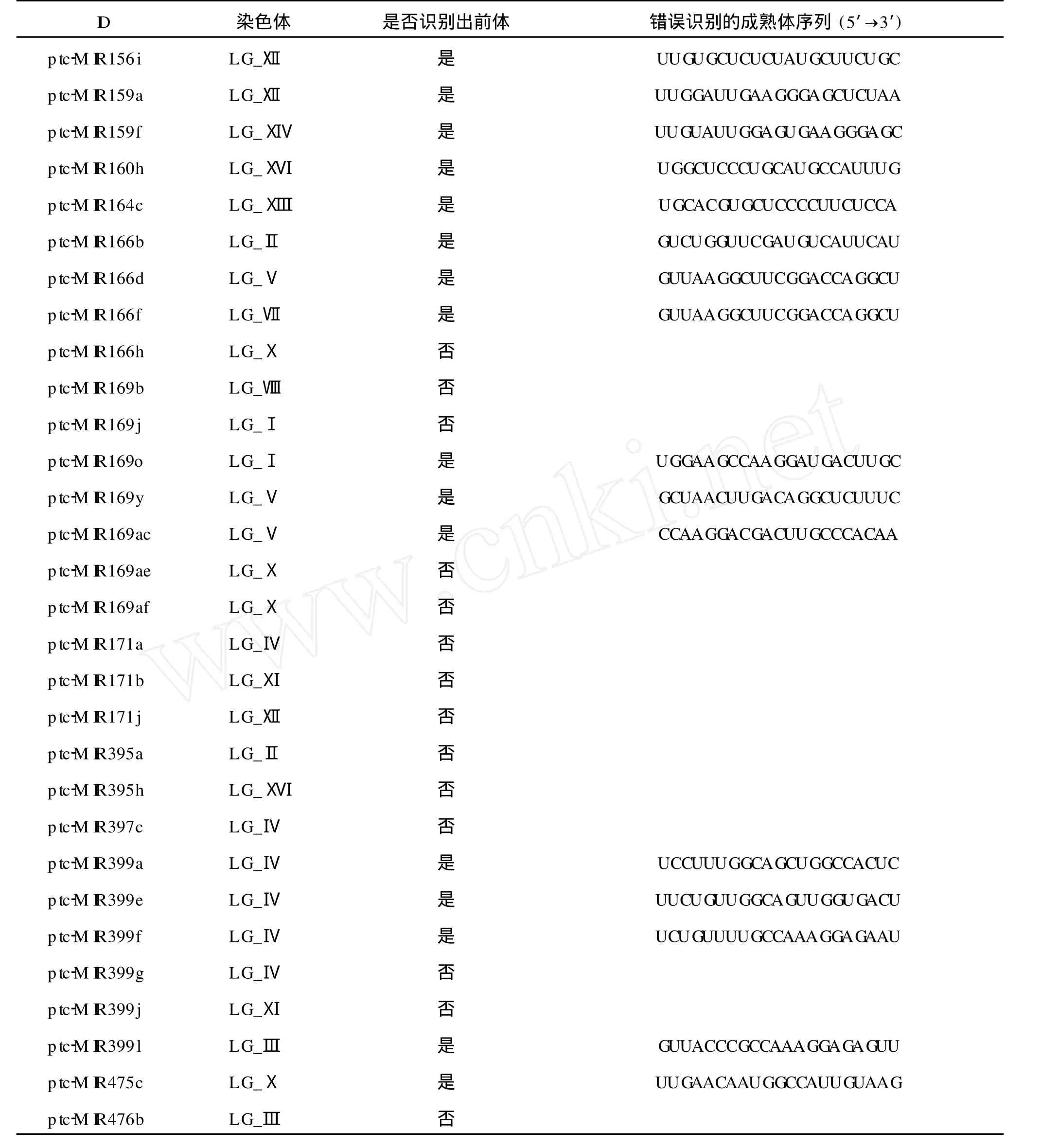
表 4 未成功识别的 m iRNATable 4 Un identif ied m iRNAs
虽然小分子的 miRNA可能以几乎任意序列存在,其前体发卡环形的二级结构和它在前体的位置却呈现出十分固定的特点,可以说,相对于序列间的相似性,miRNA更具备结构上的相似性[2].改进M iRfilter的时候,更注重加强 miRNA结构信息的描述.在 23个训练参数中,有 4个序列参数和 19个结构参数;在 12个用于打分的变量中,也只有 2个与序列信息有关.对 miRNA结构特征的关注使得M iRfilter可以在不搜索同源片段的情况下仍然表具有良数好据的预测精度.

Table 5 False positivem iRNAs

表 6 M iRf ilter系统预测精度Table 6 Pred iction accuracy of M iRf ilter
虽然制定了严格的筛选标准来降低假阳性率,仍然会得到大量的预测结果,为了确认其中的 miRNA,需要进行实验验证.目前,miRNA的实验检测方法主要有:RNA印迹 (Northem blot)、实时荧光定量 PCR(real-time PCR)、芯片技术 (microarray)等[16].每种方法都有其优缺点,可以相互结合进行检测.
miRNA的确定需要计算预测和实验检测共同完成.像M iRfilter这样的计算预测工具可以从海量的数据中筛选出合理的潜在对象,弥补实验方法效率低、成本高的缺点,已然成为实验检测不可或缺的前提条件.
[1] 华友佳,肖华胜.microRNA研究进展 [J].生命科学,2005,17(3):278-281.
[2] 金由辛.核糖核酸与核糖核酸组学[M].北京:科学出版社,2005:106-133.
[3] 王磊,范云六.植物微小 RNA(microRNA)研究进展[J].中国农业科技导报,2007,9(3):18-23.
[4] 李培旺,卢向阳,李昌珠,等.植物 microRNA s研究进展 [J].遗传,2007,29(3):283-288.
[5] DUGAS D V,BARTEL B.M icroRNA regulation of gene expression in plants[J]. Current Opinion in Plant Biology,2004,7(5):512-520.
[6] 侯妍妍,应晓敏,李伍举.M icroRNA计算发现方法的研究进展[J].遗传,2008,30(6):687-696.
[7] GLAZOV E A,COTTEE P A,BARRISW C,et al.A microRNA catalog of the developing chicken embryo identified by a deep sequencing app roach[J].Genome Research,2008,18(6):957-964.
[8] RITCHIEW,THEODULE FX,GAUTHERETD.M ireval:a web tool for simple microRNA p rediction in genome sequences[J].Bioinformatics,2008,24(11):1394-1396.
[9] WANG X J,REYES J L,CHUA N H,et al.Prediction and identification of Arabidopsis thaliana microRNAs and theirmRNA targets[J].Genome Biology,2004,5(9):R65.
[10] XUE C H,L I F,HE T,et al.Classification of real and pseudo microRNA p recursors using local structuresequence features and support vector machine[J].BMC Bioinformatics,2005,6:310.
[11] HERTEL J,STADLER P F.Hairp ins in a Haystack:recognizing microRNA precursors in comparative genomics data[J].Bioinformatics,2006,22(14):197-202.
[12] 张玉滨.基于生物信息学方法预测 microRNA的研究[D].上海:上海大学,2007:36-49.
[13] 陈薇,谭军,何晨.植物 miRNAs前体的生物信息分析[J].重庆邮电学院学报:自然科学版,2006,18(6):803-808.
[14] YOUSEFM,JUNG S,SHOWE L C,et al.Learning from positive examples when the negative class is undetermined-microRNA gene identification [J].Algorithms for Molecular Biology,2008,3:2.
[15] 甘四明,苏晓华.林木基因组学研究进展 [J].植物生理与分子生物学学报,2006,32(2):133-142.
[16] 王旭丹.M icroRNA检测方法的发展现状[J].国际内科学杂志,2007,34(11):679-682.
(编辑:刘志强)
Pred iction of Populus trichocarpa m iRNAs w ith Im proved M iRf ilter System
ZHAO Jie-yuan, GONG Yun-lu, WANG Yi-fei
(College of Sciences,Shanghai University,Shanghai200444,China)
O 224
A
1007-2861(2010)04-0397-07
10.3969/j.issn.1007-2861.2010.04.014
2009-02-20
国家自然科学基金资助项目 (30871341);上海市重点学科建设资助项目 (S30104);上海市教委重点学科建设资助项目(J50101)
王翼飞 (1948~),男,教授,博士生导师,研究方向为计算分子生物学.E-mail:yifei_wang@staff.shu.edu.cn
猜你喜欢
故事作文·高年级(2023年3期)2023-05-30 19:00:18
天津医科大学学报(2021年1期)2021-12-05 11:11:05
今日农业(2021年11期)2021-08-13 08:53:24
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
分析测试学报(2015年5期)2016-01-13 06:18:50
中成药(2014年11期)2014-02-28 22:30:08
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
茶叶通讯(2014年2期)2014-02-27 07:55:40
世界科学(2013年6期)2013-03-11 18:09:33
