
RP-HPLC测定鹿尾鞭酒中淫羊藿苷的含量
2010-10-16 02:34姜涛孙晶晶
中国现代中药 2010年9期
姜涛,孙晶晶
(吉林省食品药品检验所,吉林 长春 130033)
RP-HPLC测定鹿尾鞭酒中淫羊藿苷的含量
姜涛*,孙晶晶
(吉林省食品药品检验所,吉林 长春 130033)
目的:建立鹿尾鞭酒中淫羊藿苷的含量测定方法。方法:使用RP-HPLC对该制剂中淫羊藿苷进行含量测定,色谱柱:Agilent HC-C18(250 mm×4.6 mm,5μm),流动相:乙腈-水(23∶77),流速:1.0 mL·min-1,检测波长:270 nm。结果:淫羊藿苷进样量在0.177 12~0.619 92μg线性关系良好(r=0.999 9),平均回收率为96.62%,RSD=1.39%(n=6)。结论:本方法可有效控制鹿尾鞭酒质量。
鹿尾鞭酒;淫羊藿苷;反相高效液相色谱法
鹿尾鞭酒是由中药鹿尾、驴肾、狗肾、人参、熟地黄、淫羊藿、鹿茸、锁阳、肉苁蓉、补骨脂(盐制)、当归、菟丝子、甘草经现代工艺加工制成的中药复方制剂。本品收载于 《卫生部药品标准·中药成方制剂》第13册,具有补肾壮阳,用于肾虚体弱,腰膝无力等症[1]。原质量标准中无定性、定量检测方法,为了更好地控制药品质量,促进合理用药,对鹿尾鞭酒中有效成分的质量控制方法进行提高成为一项比较迫切的工作。淫羊藿为历代常用的补益中药,已有研究表明淫羊藿苷具有抗氧化、抗衰老、抗炎镇痛、免疫调节、抗菌抗病毒、预防心脑血管疾病等多种生理功能[2],是鹿尾鞭酒中淫羊藿的有效成分,可作为定量标准物质。本实验采用RPHPLC对淫羊藿苷进行含量测定,为该制剂质量控制提供一种简便、准确的方法。
1 仪器与试药
岛津LC-20AT高效液相色谱仪;AE-163电子天平(瑞士梅特勒);岛津UV-2201紫外-可见分光光度计;淫羊藿苷对照品(中国药品生物制品检定所,批号0737-200111,供含量测定用)。乙腈为色谱纯,其他试剂均为分析纯。鹿尾鞭酒(辽源誉隆亚东药业有限责任公司,批号050702,061201,061202)。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent HC-C18(250 mm×4.6 mm,5μm);流动相:乙腈-水(23∶77);流速:0.8 mL·min-1;柱温:35℃;检测波长:270 nm;理论板数按淫羊藿苷峰计算,应不低于5 000。
2.2 对照品溶液的制备
取淫羊藿苷对照品适量,精密称定,加甲醇制成每1 mL含淫羊藿苷0.045 mg的溶液,即得。
2.3 供试品溶液的制备
精密量取本品25 mL,水浴浓缩至约5 mL,置D101大孔吸附树脂柱(内径1.5 cm,长12 cm)上,以1.5mL·min-1的流速,用水100 mL洗脱,弃去水液,再用70%乙醇洗脱,弃去初洗脱液5~8 mL,收集洗脱液100mL,蒸干,用70%乙醇溶解并转移至10 mL量瓶中,加70%乙醇稀释至刻度,摇匀,滤过,即得。
2.4 阴性对照试验
为进一步考察实验设计的合理性,取不含淫羊藿的阴性对照样品适量,依上述色谱条件测定,结果,在所得色谱图中在与淫羊藿苷峰相应的保留时间附近无干扰峰检出,说明本法是合理可行的,见图1。
2.5 标准曲线的制备
精密称取淫羊藿苷对照品适量,加甲醇制成每1 mL含0.044 28 mg的溶液,分别精密吸取4,6,8,10,12,14μL,注入液相色谱仪,测定,以对照品的进样量为横坐标,色谱峰面积值为纵坐标,得回归方程Y=2 661 214X-828 2,r=0.999 9。结果表明,淫羊藿苷进样量在0.177 12~0.619 92μg有良好的线性关系。
2.6 精密度试验
精密吸取对照品溶液10μL,注入液相色谱仪,重复进样6次,测定其色谱峰面积值,RSD=0.2%,结果表明所用仪器具良好的精密性。
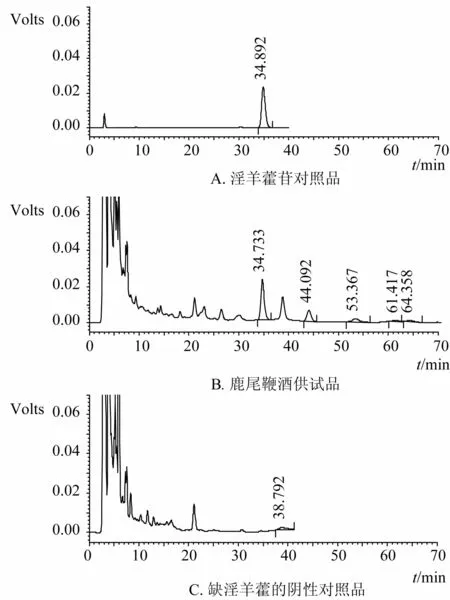
图1 鹿尾鞭酒对照品及供试品HPLC图
2.7 稳定性试验
取同一供试品溶液,在0,2,4,8,16,24 h按含量测定下色谱条件测定,RSD=0.8%,结果表明24 h内供试品溶液中淫羊藿苷的含量基本稳定。
2.8 重现性试验
取同一供试品(批号050702),依含量测定方法独立测定6次,计算供试品中淫羊藿苷的平均含量为0.016 9 mg·mL-1,RSD=1.2%。结果表明,该方法重现性良好。
2.9 加样回收率试验
精密称取同法测定已知含量的供试品(批号050702,含量为0.016 9 mg·mL-1)6份,精密加入淫羊藿苷对照品溶液(0.073 8 mg·mL-1)2.5 mL,依法测定,计算回收率,结果见表1。
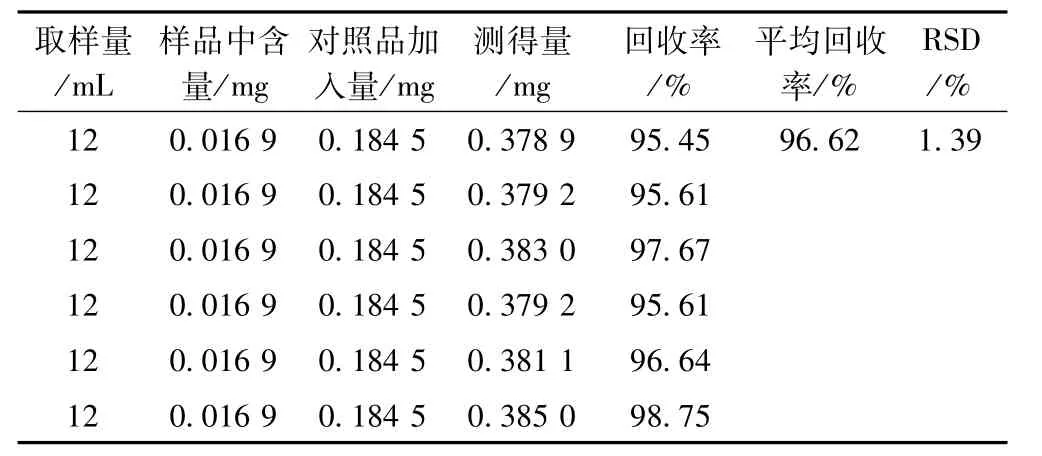
表1 淫羊藿苷加样回收率
2.10 样品含量测定
取鹿尾鞭酒供试品050702,061201,061202 3批,按2.3方法制备后分别进样,结果分别为0.017 4,0.012 2,0.012 0 mg·mL-1(n=2)。
3 讨论
3.1 耐用性试验考察
采用Agilent HC-C18(4.6 mm×250 mm,5μm)、Apollo C18(4.6 mm×250 mm,5μm)、Agela C18(4.6 mm×250 mm,5μm)3根不同品牌色谱柱进行试验,并参考 《中国药典》2010年版一部 “淫羊藿”含量测定项下要求,确定本品理论板数按淫羊藿苷峰计算应不低于5 000。
3.2 测定波长的选择
取淫羊藿苷对照品甲醇溶液,置岛津UV-2201紫外-可见分光光度计上,在波长范围200~320 nm测定,结果其最大吸收波长为270 nm,与 《中国药典》2010年版一部淫羊藿项下的检测波长相符。故选择270 nm为测定波长。
3.3 流动相选择
本品为酒剂,药味组分多,成分复杂,相互干扰较多。曾参考 《中国药典》淫羊藿药材中淫羊藿苷的含量测定方法及其他文献中淫羊藿苷的含量测定方法,均采用高效液相色谱法测定淫羊藿苷的含量[3-5],选用乙腈-水(30∶70)为流动相,分离效果不理想,调整比例为23∶77后,分离完全,故选择乙腈-水(23∶77)为流动相。
3.4 层析条件的考察
D101大孔吸附树脂柱层析条件的考察,是为了除去制剂中糖等水溶性杂质。实验将上样后的水洗脱液蒸干,以甲醇定容至10 mL量瓶中,测定其色谱图,结果无淫羊藿苷峰,表明该洗脱过程中,测定组分基本无损失,同时比较了收集70%乙醇洗脱液100,120 mL淫羊藿苷的量,结果100 mL和120 mL基本相同,故选择了100 mL为本法的收集量。
本实验为鹿尾鞭酒中淫羊藿苷的含量测定提供方法学依据,建立的方法科学可行,具有很好的重现性,为鹿尾鞭酒的质量标准提高提供了科学依据。
[1]卫生部.卫生部药品标准·中药成方制剂(第13册)[S].1997:189.
[2]马金秋,马向前,张晓宇.淫羊藿黄酮抗运动性疲劳作用机制研究[J].中国新药杂志,2009,18(6):553-559.
[3]国家药典委员会.中国药典(一部)[S].北京:中国医药科技出版社,2010:306-308.
[4]王蕊,张峰,李长富,等.HPLC测定前列舒乐胶囊中淫羊藿苷的含量 [J].中国现代中药,2006,8(4):14-16.
[5]陈刚,彭艳,陈熙佳.高效液相色谱法测定男宝胶囊中淫羊藿苷含量 [J].中国药业,2009,18(1):13-14.
Determination of Icariin in Luweibian Wine by RP-HPLC
Jiang Tao,Sun Jingjing
(Jilin institute for food and drug control,Changchun Jilin130033,China)
Objective:To establish a determination method of Icariin in Luweibian wine.Methods:The Icariin was determined by RP-HPLC.Themobile phase was acetonitrile and water,the flow rate was1.0mL·min-1,the UV detection wavelength was at270 nm;Results:The linearity was obtained over the range of0.177 12~0.619 92μg(r=0.999 9)for Icariin.The average recovery was 96.62%,RSD was 1.39% (n=6).Conclusion:The method can be used for the quality control of Luweibian wine.
Luweibian wine;Icariin;RP-HPLC
*姜涛,E-mail:tnt_tao@sina.com
2010-04-19)
猜你喜欢
江西中医药(2022年8期)2022-08-22
青岛大学学报(医学版)(2022年3期)2022-08-05
煤化工(2022年3期)2022-07-08
色谱(2021年7期)2021-06-07
环球中医药(2021年3期)2021-04-07
中成药(2018年12期)2018-12-29
中国中医药现代远程教育(2018年22期)2018-02-09
中成药(2017年4期)2017-05-17
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
