
基因组规模代谢网络模型构建及其应用
2010-10-11 02:11刘立明陈坚
生物工程学报 2010年9期
刘立明,陈坚
江南大学 食品科学与技术国家重点实验室,无锡 214122
代谢工程
基因组规模代谢网络模型构建及其应用
刘立明,陈坚
江南大学 食品科学与技术国家重点实验室,无锡 214122
微生物制造产业的发展迫切需要进一步提高认识、设计和改造微生物细胞代谢的能力,以推动工业生物技术快速发展。随着微生物全基因组序列等高通量数据的不断积聚和生物信息学策略的持续涌现,使全局性、系统化地解析、设计、调控微生物生理代谢功能成为可能。而基于基因组序列注释和详细生化信息整合的基因组规模代谢网络模型 (GSMM) 构建为全局理解和理性调控微生物生理代谢功能提供了最佳平台。以下在详述GSMM的应用基础上,描述了如何构建一个高精确度的GSMM,并展望了未来的发展方向。
基因组规模代谢网络,微生物制造,构建,应用
Abstract:The exploitation of microbial manufacture process (MMP) in industrial biotechnology requires a comprehensive understanding and an efficient modification of microorganism physiology. The availability of genome sequences and accumulation of-omics data allow us to understand of microbial physiology at the systems level, and genome-scale metabolic model (GSMM)represents a valuable framework for integrative analysis of metabolism of microorganisms. Genome scale metabolic models are reconstructed based on a combination of genome sequence and the more detailed biochemical knowledge, and these reconstructed models can be used for analyzing and simulating the operation of metabolism in response to different perturbations. Here we describe the reconstruction protocols for GSMM in further detail and provide the perspective of GSMM.
Keywords:genome scale metabolic model, microbial manufacture process, reconstruction, application
1 后基因组时代的微生物制造
面对石油等不可再生的化石资源严重短缺并日渐枯竭,以及化学加工对环境污染日益加深的现实,世界经济发展模式逐渐从不计消耗、牺牲环境的化石经济模式转向以可再生的碳水化合物为原料、利用生物细胞或酶的生物催化功能进行大规模物质加工与转化的生物经济模式,从而在源头上解决资源短缺与环境污染问题[1]。生物经济的重要支柱之一是微生物制造。然而,由于微生物细胞或酶具有为自身服务的本能,其代谢过程处于最经济的状态,使得其工作效率成为制约微生物制造产业发展的瓶颈。因此,迫切需要通过代谢工程等策略以进一步提高认识、设计和改造微生物细胞代谢功能的能力,推动微生物制造技术的进步,支持工业可持续发展。但是,以往一系列的代谢工程改造策略,包括减弱反馈抑制、消除竞争途径、过量表达主要合成途径、提升细胞转运能力、引入全新代谢途径以修饰微生物代谢途径中的某一个或几个基因,不一定能获得预期的代谢通量增大或降低的效果[2]。出现这一状况的原因在于缺乏对微生物代谢网络全局调控和表达调控网络的充分理解。
另一方面,随着基因组及其相关技术的发展,几乎所有重要工业微生物模式种的全基因组都已经或即将公布,微生物制造研究进入了后基因组时代。后基因组时代的微生物制造具有以下特点:1) 源于微生物基因组的蛋白质组数据的积聚,使认识、理性设计、定向改变酶的性能更为容易;2) 转录组学、代谢组学等数据的积聚有助于理解、提高工业微生物抵御微生物制造过程中的恶劣环境;3) 从对微生物中个别基因或蛋白质功能的局部性研究,转移到以细胞内全部基因、mRNA、蛋白质以及代谢产物为研究对象的工业微生物生理研究;4) 基于生物信息学的基因组规模基因调控网络、信号转导网络、蛋白质相互作用网络和代谢网络的构建、模拟与分析,逐步将代谢工程推入更为理性的系统代谢工程时代[3];5) 采用合成生物学的技术精确控制代谢途径、合成目的蛋白,构建“人工细胞”,显著提高微生物制造效率。前已述及,由于基因与蛋白质倾向于成组地通过网状相互作用而影响微生物细胞功能,因此对工业微生物生理功能的理解和全局调控的研究必须构建并分析其相互作用的网络。这些分子和基因相互作用网络包括基因调控网络、信号转导网络、蛋白质相互作用网络和代谢网络等。其中,基于基因组注释的代谢网络把微生物细胞内所有生化反应构建为一个网络模型,反映了所有参与代谢过程的化合物之间以及所有催化酶之间的相互作用。基于基因组数据构建的特定微生物代谢网络,及其结构和功能的分析,为从全局规模上深刻认识和高效、定向调控微生物生理功能奠定了坚实基础,从而为代谢工程的发展创造了前所未有的机遇[4-5]。
2 基因组规模代谢网络模型的应用
基因组规模代谢网络模型 (Genome scale of metabolic network model,GSMM) 包含了其所给定微生物内部发生的绝大部分生化反应。BIGG (http://bigg.ucsd.edu/) 数据库表明,原核微生物的代谢网络包含了1 000多个代谢物、1 200多个基因和2 000多个生化反应 (以E. coli为例);而真核微生物代谢网络模型包括1 200多个代谢物、1 000多个基因和1 500多个生化反应 (以S. cerevisiae为例)。基因组规模代谢网络模型一般能注释约 6%~15% (真核微生物) 和25% (原核微生物)的开放阅读框 (ORF)。因此,基因组规模代谢网络模型能提供一个高效平台以全局理解微生物生理代谢功能 (图 1)。主要应用于以下4个方面[6](表1):1) 分析代谢网络属性;2) 预测和分析微生物生长表型;3) 基于模型的组学数据阐释[7];4) 系统代谢工程。
通过GSMM,可以从代谢物和代谢流两个水平上充分理解微生物的代谢特性。在代谢物水平上,将基因组规模代谢网络模型中的代谢物浓度范围与GC-MS实验所测定的S. cerevisiae中间代谢产物真实浓度进行比较,发现有52种代谢物浓度存在差异,并结合转录组数据的分析,找到了调控S. cerevisiae代谢功能的关键因子[31]。另一方面,整合 C13同位素标记实验数据,GSMM用以预测难以测定的中间代谢物浓度范围、反应进行方向、Gibbs自由能范围和潜在的调控反应等[32]。此外,GSMM还可预测代谢反应的中间代谢物之间的共生与保守关系[33]。在代谢流水平上[34-35],通过分析不同环境下不同代谢反应上代谢流分布的情况,发现微生物细胞是由少数承担着绝大部分代谢通量的骨干代谢途径和大量附属代谢途径组成。与此相类似,借助GSMM还可发现不同代谢反应之间的关系和依赖性,鉴别新代谢途径,发现任何状态下不承担任何代谢流的生化反应,研究必需代谢途径等。

表1 基因组规模代谢网络模型的应用Table 1 Application of genome scale metabolic model

续表1
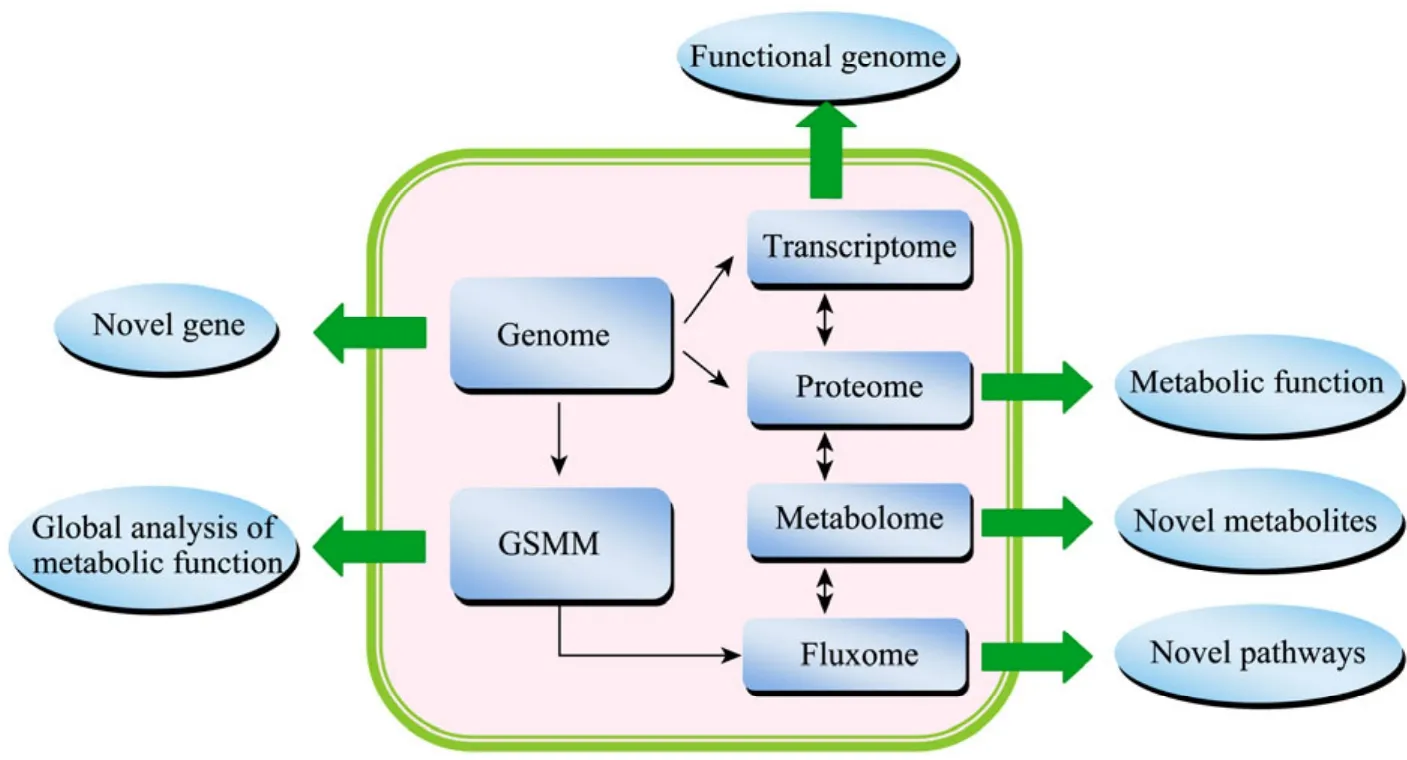
图1 基因组规模代谢网络模型是系统理解微生物代谢功能的平台Fig.1 GSMM offer a platform to globally and comprehensive understanding the microbial physiology.
微生物细胞内的代谢调控发生在基因-基因、基因-蛋白质、蛋白质-蛋白质、蛋白质-代谢物以及物理间相互作用等水平上。通过与其他组学数据,如转录组、蛋白质组等的整合,GSMM用以充分理解微生物细胞内的调控机制。通过比较不同碳源下恒化培养时S. cerevisiae的代谢流分布和转录组数据,研究人员重构了一个由348个代谢基因和55个调节子组成的代谢调节网络,揭示了S. cerevisiae的代谢调节机制[36]。借助代谢流分析,研究人员深度解析了E. coli全局代谢调控蛋白ArcA、ArcB、Cra、Crp、Cya、Fnr和Mlc对葡萄糖代谢的影响[37]。pckA和pps的表达水平因csrA的删除而受到抑制,从而导致氨基酸合成前体磷酸烯醇式丙酮酸的胞内浓度不断下降[38]。相反,若过量表达pps则能有效提高苯丙氨酸的产量[39]。进一步深入比较分析E. coli基因组规模代谢网络模型的代谢通量和转录组数据发现,代谢相关基因的表达水平及通过酶节点的代谢通量不仅具有相同的表达模式,还受相同的转录调控子所调控。
由于 GSMM 是由一系列用于合成微生物生长所必需的能量和前体代谢物的生化反应所组成,因此,GSMM广泛应用于定性与定量预测不同代谢或环境搅动条件下细胞生长的表型。包括:1) 采用代谢流平衡分析 (Flux balance analysis,FBA),预测不同营养物吸收速率、代谢副产物合成速率和细胞生长速率之间的全局性关系;2) 预测不同类型培养基上细胞产率和生长比速率;3) 基于基因-蛋白质-反应 (Gene-protein-reaction,GPR) 原则预测基因删除对细胞生长表型的影响;4) 预测并构建微生物生长 (或合成目标代谢产物) 必需基因集;5) 定量预测不同环境或代谢搅动下微生物生长表型。
建立在充分理解微生物细胞代谢功能基础之上,借助重组DNA技术有目的地设计和改造微生物细胞内代谢网络和表达网络,以提高微生物代谢效率的代谢工程技术,有效地克服了传统育种手段的突变非定向性和设计非理性的缺点,广泛应用于微生物制造领域以提高目标代谢产物生产效率。随着基因组规模技术的发展,代谢工程逐渐转移到系统代谢工程,也即从全局的角度研究和调控微生物细胞的代谢与调控网络。基于此,GSMM为代谢工程提供了研究目标微生物代谢功能的有效工具:1)根据代谢能力或最大理论转化率选择宿主菌;2) 根据代谢流分布确定微生物细胞内瓶颈反应,确定代谢工程目标,发展代谢工程策略;3) 预测和评价代谢改造后突变株的代谢表型。
3 基因组规模代谢网络模型的构建
3.1 GSMM构建所需数据库与软件
基因组规模代谢网络模型构建过程是建立基因-蛋白质-生化反应之间联系的过程,涉及到基因组、蛋白质和酶、中间代谢产物、生化反应、代谢途径等诸多方面的数据。所需数据库和计算工具的介绍发表在每年的“Nucleic Acids Research”(NAR database issue) 第1期上。构建过程所需的数据库和工具详细列于表 2,主要分为以下几类:1) 基因组数据库。收集了来自全球的核酸序列主要包括EMBL数据库 (由欧洲生物信息学研究所管理)、GenBank (由美国国家生物技术信息中心管理) 和DDBJ (由日本三岛信息生物学中心管理)。上述3个数据库虽然格式有所不同,但内容完全相同。一般而言,能从上述3个数据库获得所给定微生物的全基因组序列 (核酸或蛋白质)。此外,一些已经测序的微生物建立了自己特有的数据库,不断更新经注释的基因组信息,如黑曲霉 (http://genome.jgi-psf.org/Aspni1/Aspni1.home.html) 的基因组数据库。这些专业数据库收集的是与明确功能类别或分类学标准相关的生物学数据。由于是经过修正和分析后产生的注释数据库,数据的精确度和验证能力更胜一筹。2) 蛋白质数据库。蛋白质数据库收集了蛋白质序列、分类、功能、结构域、蛋白质模体 (Motif)、亚细胞定位等信息,而酶学数据库则收集了酶的分类、功能、催化反应、标准命名以及酶相关特性,如温度、Km值、pH等以及与之相关的事实文献。3) 代谢物数据库。GSMM构建中所涉及的中间代谢产物的信息,如代谢物名称、化学式、InChI、化学结构、生物学功能等从ChEBI、PubChem和KEGG等3个数据库中获得。4) 代谢途径数据库。代谢途径数据库提供了微生物细胞内代谢反应的名称、代谢物、辅因子、反应方向、代谢途径等信息。最为典型的代谢途径数据库是 EcoCyc Ontology和KEGG。EcoCyc Ontology收录了E. coli基因和其他一些相关微生物代谢途径,同时通过途径/基因组的用户界面可视化地展现了细菌染色体内的基因轮廓、单个生化反应和整个生化代谢途径。KEGG收录了所有与途径相关的数据,包括途径中相关的基因和化合物的目录[40]。KEGG还提供了途径图谱、直系族表、分子目录、基因组图谱和基因目录等数据。这些数据不仅相互之间互为链接并与其他相关数据库进行链接,且能通过整合的数据库进行检索。5) 文献事实数据库[41]。随着高通量技术手段的不断发展,生物学实验手段和研究方法均发生了巨大变化,由此带来实验数据以指数方式增长。以隶属于美国国家生物技术信息中心的 MEDLINE为例,该数据库收集了全世界 4 800多种生物学及医学杂志所发表的1.3亿余篇文献,并以每月几万篇的速度增长。文献事实数据库为 GSMM构建提供以下信息[42-43]:(I) 给定基因的名称、序列、亚细胞位置、转录调节以及相应生理功能;(II) 蛋白质序列、结构域、功能模块、结构特性和功能;(III) 酶的名称、EC号、生理与功能参数、结构特征、稳定性、抑制剂或激活剂、应用以及与特定微生物相关的信息;(IV) 生化反应底物与产物、辅因子、单个生化反应和代谢途径;(V) 代谢网络与蛋白质相互作用网络;(VI) 转录调控、底物和代谢产物的跨膜转运、代谢产物的种类;(VII) 细胞组成成分、生理参数和特定微生物的相关生化参数 (如培养基组分等)。6) 模型构建的软件:构建过程使用的软件主要是 Matlab,需要构建者熟悉Matlab编程;另一类是模型可视化软件,主要是Illustrator和CellDesigner。后者可与其他组学数据如转录组进行交互阐释。

表2 基因组规模代谢网络模型构建所需数据库Table 2 Database and software for reconstruction of genome scale metabolic network

续表2

图2 基于基因组注释的粗略代谢网络构建步骤Fig.2 Elements and reconstruction protocol of the draft model based on the genome annotation.
GSMM构建一般分为5个阶段[44]:1) 基因组注释与粗略模型的获得;2) 构建精细代谢模型;3) 数学模型的转换;4) 识别与填补代谢空洞 (Metabolic gaps);5) 模拟与可视化。
3.2 基于基因组注释的粗略代谢模型构建
构建 GSMM 需要从基因组数据库获取目标微生物的全基因组序列,再通过 Blastp、KAAS等方法对全基因组进行注释,以建立基因-蛋白质反应之间的关系。从基因组注释到构建粗略模型过程中需要收集的信息详列于图 2中。在将基因组序列注释为蛋白质 (酶的名称和EC号) 进而到生化反应的过程中需要注意以下问题:1) 有些蛋白质虽能注释为酶的催化活性但没有EC号;2) 有些蛋白质功能注释过程中仅提供GO,而没有分配系统的EC号;3)同一个酶能催化不同底物为不同的产物;4) 同工酶虽由不同基因编码,但只能催化同一个反应;5) 不同基因编码复合酶所催化的不同反应,但其所处的亚细胞位置不尽相同。上述问题的解决依赖于进一步的文献挖掘。
3.3 基于手工修正的精细代谢网络
通过基因组注释得到粗略代谢网络(以Excel形式存储)的基础上,需要添加一系列基因组注释过程中无法提供的生化反应以降低dead-end代谢物的数量 (图3):自发反应 (Spontaneous reactions);跨膜运输反应;胞内细胞器间运输反应;交换反应(Exchange reactions)。但若代谢网络模型中包含过多的运输反应,会导致很多无效循环的发生。因此,在基于文献挖掘添加上述反应的基本原则是:添加确实需要的生化反应。同时,评价一个基因组规模代谢网络模型成功与否的标准是该模型能否在给定培养条件下真实预测细胞生长,因此,GSMM需包括与细胞生长的相关反应:蛋白质合成反应;细胞壁组分合成反应;脂类合成反应;核苷酸类物质合成反应;脱氧核苷酸类物质合成反应;细胞生长和细胞维持所需ATP等。紧随其后,需采用人工核查的方式检查每一个生化反应,包括:单个反应所需底物、产物、辅因子的种类、分子式、化学计量学平衡等;基于吉布斯自由能的生化反应方向;生化反应的亚细胞定位、子系统;源自文献挖掘的实验证据。
3.4 转换为数学模型
GSMM构建的第3个阶段是在安装COBRA和SBML工具箱后,利用Matlab将精细代谢网络转换为数学模型,以供后续验证和模拟使用。然而,采用 GSMM 模拟细胞生长或目标代谢产物的合成始终与实验数据存在一定的差距,其原因在于模拟代谢流的分布缺乏一定的约束条件。为了使GSMM模拟结果更接近微生物真实的代谢,在GSMM模拟过程中通常添加一定的约束条件:物理-化学约束;拓扑约束;环境条件约束;代谢调控约束。GSMM中最常用的约束条件是物理-化学约束和环境条件约束 (图 4):代谢通量平衡 (S⋅v=0);能量平衡 (ΔE=0);酶催化或细胞转运的能力 (vi≤vmax) 以及热动力学参数 (0≤vmin)。
3.5 评估基因组规模代谢网络模型

图3 精细GSMM需要添加的反应与示例Fig.3 Added reactions to GSMM during the manual reconstruction refinement.

图4 GSMM模拟的约束条件Fig.4 Physic-chemical and environmental conditions constraints for GSMM simulation.
将代谢网络模型转换为数学模型后,随后的工作是采取一系列步骤评估与修正所构建的代谢网络模型:1) 从碳、氮、硫、磷、氢和电荷等方面查找和修补物质或电子不平衡的生化反应;2) 查找代谢末端 (Metabolic dead ends) 以鉴定代谢漏洞(Metabolic gaps),进而通过基因注释和文献挖掘寻找相应生化反应修补相应的代谢漏洞;3) 添加缺失的交换反应(Exchange reactions) 以及设置交换反应的约束条件;4) 基于热力学特性、ATP和 NADH合成与利用调整生化反应方向;5) 鉴定孤儿反应(Orphan reaction),调整或删除引起代谢冗余的生化反应。在填补代谢漏洞的过程中,需要注意的事项是新添加到代谢网络中的生化反应可能会导致新漏洞出现。因此,当加入新的反应时,应确保所有代谢物能连接到已构建好的代谢网络。评价GSMM构建精确性还包括以下指标:是否在不同培养基和给定的培养条件下合成细胞生长所需的前体物质;典型培养基上能否生长;不同培养基上的细胞浓度和生长速度的比较;合成目标代谢产物的能力及其合成速率。
3.6 GSMM模拟和可视化
4 展望
通过GSMM构建与模拟,研究人员能在不同组学水平上全局性、定量化理解微生物细胞的生理代谢功能,进而设计代谢工程策略,获取所需的生理表型,提高微生物制造的效率。然而,由于难以全面获取微生物细胞酶动力学相关数据和相关的调控机制,导致不能全面正确地解析和调控微生物细胞的代谢功能,进而提高工业生物过程的效率。为了更好地理解和整合微生物细胞内代谢调节机制和酶动力学知识,因此需要设计和应用更为新颖的生物信息学工具与方法。因此,为了更好地在全局性解析微生物生理功能的基础上,发展高效代谢工程策略,定向改造微生物菌株为工业生物技术服务,今后的研究应注重以下两个方面:构建更为全面、准确的包含调控机制和动态动力学参数的GSMM;发展新的模拟计算方法和约束条件,以更加准确地模拟真实的微生物细胞代谢。
REFERENCES
[1] Otero JM, Nielsen J. Industrial systems biology.Biotechnol Bioeng, 2009, 105: 439−460.
[2] Feist AM, Palsson BO. The growing scope of applications of genome-scale metabolic reconstructions usingEscherichia coli.Nat Biotechnol, 2008, 26: 659−667.
[3] Park JH, Lee SY. Towards systems metabolic engineering of microorganisms for amino acid production.Curr Opin Biotechnol, 2008, 19: 454−460.
[4] Patil KR, Akesson M, Nielsen J. Use of genome-scalemicrobial models for metabolic engineering.Curr Opin Biotechnol, 2004, 15: 64−69.
[5] Feist AM, Palsson BO. The growing scope of applications of genome-scale metabolic reconstructions usingEscherichia coli.Nat Biotechnol, 2008, 26: 659−667.
[6] Durot M, Bourguignon PY, Schachter V. Genome-scale models of bacterial metabolism: reconstruction and applications.FEMS Microbiol Rev, 2009, 33: 164−190.
从2003年的孙志刚案最终导致废除收容审查制度,到如今于海明案让我们对长期处于“认定模糊地带”的法律概念——正当防卫,有了更深刻的理解,我们相信,每一位有着最朴素正义观的人民群众,在一起起个案中共同提升了法律素养和法治意识;我们更相信,法律从来不是冰冷的,而中国司法也必将在追求法律效果和社会效果的统一中,温暖而坚定地前行!如同习近平总书记说,“努力让人民群众在每一个司法案件中都感受到公平正义”!
[7] Hanisch D, Zien A, Zimmer R,et al. Co-clustering of biological networks and gene expression data.Bioinformatics, 2002, 18(suppl 1): S145−S154.
[8] Reed JL, Palsson BO. Genome-scalein silicomodels ofE. colihave multiple equivalent phenotypic states:assessment of correlated reaction subsets that comprise network states.Genome Res, 2004, 14: 1797−1805.
[9] Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models.Metab Eng, 2003, 5: 264−276.
[10] Schilling CH, Palsson BO. Assessment of the metabolic capabilities ofHaemophilus influenzaeRd through a genome-scale pathway analysis.J Theor Biol, 2000, 203:249−283.
[11] Klamt S, Stelling J. Two approaches for metabolic pathway analysis?Trends Biotechnol, 2003, 21: 64−69.
[12] Burgard AP, Nikolaev EV, Schilling CH,et al. Flux coupling analysis of genome-scale metabolic network reconstructions.Genome Res, 2004, 14: 301−312.
[13] Nikolaev EV, Burgard AP, Maranas CD. Elucidation and structural analysis of conserved pools for genome-scale metabolic reconstructions.Biophys J, 2005, 88: 37−49.
[14] Imielinski M, Belta C, Halasz A,et al. Investigating metabolite essentiality through genome-scale analysis ofEscherichia coliproduction capabilities.Bioinformatics,2005, 21: 2008−2016.
[15] Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic byproduct secretion in wild-typeEscherichia coliW3110.Appl Environ Microb, 1994, 60: 3724−3731.
[16] Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks.Proc Natl Acad Sci USA, 2002, 99: 15112−15117.
[17] Shlomi T, Berkman O, Ruppin E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations.Proc Natl Acad Sci USA, 2005, 102:7695−7700.
[18] Imielinski M, Belta C. Exploiting the pathway structure of metabolism to reveal high-order epistasis.BMC Syst Biol,2008, 2: 40.
[19] Sauer U. Metabolic networks in motion: 13C-based flux analysis.Mol Syst Biol, 2006, 2: 6.
[20] Herrgard MJ, Fong SS, Palsson BØ. Identification of genome-scale metabolic network models using experimentally measured flux profiles.PLoS Comput Biol,2006, 2: e72.
[21] Schuetz R, Kuepfer L, Sauer U. Systematic evaluation of objective functions for predicting intracellular fluxes inEscherichia coli.Mol Syst Biol, 2007, 3: 119.
[22] Oh YK, Palsson BO, Park SM,et al. Genome-scale reconstruction of metabolic network inBacillus subtilisbased on high-throughput phenotyping and gene essentiality data.J Biol Chem, 2007, 282: 28791−28799.
[23] Henry CS, Broadbelt LJ, Hatzimanikatis V.Thermodynamics-based metabolic flux analysis.Biophys J, 2007, 92: 1792−1805.
[24] Patil KR, Nielsen J. Uncovering transcriptional regulation of metabolism by using metabolic network topology.Proc Natl Acad Sci USA, 2005, 102: 2685−2689.
[25] Akesson M, Forster J, Nielsen J. Integration of gene expression data into genome-scale metabolic models.Metab Eng, 2004, 6: 285−293.
[26] Becker SA, Palsson BO. Context-specific metabolic networks are consistent with experiments.PLoS Comput Biol, 2008, 4: e1000082.
[27] Barrett CL, Palsson BO. Iterative reconstruction of transcriptional regulatory networks: an algorithmic approach.PLoS Comput Biol, 2006, 2: e52.
[28] Alper H, Miyaoku K, Stephanopoulos G. Construction of lycopene-overproducingE. colistrains by combining systematic and combinatorial gene knockout targets.Nat Biotechnol, 2005, 23: 612−616.
[29] Pharkya P, Burgard AP, Maranas CD. OptStrain: a computational framework for redesign of microbial production systems.Genome Res, 2004, 14: 2367−2376.
[30] Lee KH, Park JH, Kim TY,et al. Systems metabolic engineering ofEscherichia colifor L-threonine production.Mol Syst Biol, 2007, 3: 149.
[31] Imielinski M, Belta C, Rubin H,et al. Systematic analysis of conservation relations inEscherichia coligenome-scale metabolic network reveals novel growth media.Biophys J,2006, 90: 2659−2672.
[32] Kummel A, Panke S, Heinemann M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data.Mol Sys Biol, 2006, 2: 10.
[33] Becker SA, Price ND, Palsson BO. Metabolite coupling in genome-scale metabolic networks.BMC Bioinformatics,2006, 7: 14.
[34] Braunstein A, Mulet R, Pagnani A. Estimating the size of the solution space of metabolic networks.BMC Bioinformatics, 2008, 9: 13.
[35] Teusink B, Wiersma A, Molenaar D,et al. Analysis of growth ofLactobacillus plantarumWCFS1 on a complex medium using a genome-scale metabolic model.J Biolog Chem, 2006, 281: 40041−40048.
[36] Herrgard MJ, Lee BS, Portnoy V,et al. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms inSaccharomyces cerevisiae.Genome Res, 2006, 16: 627−635.
[37] Perrenoud A, Sauer U. Impact of global transcriptional regulation byArcA,ArcB,Cra,Crp,Cya,Fnr, andMlcon glucose catabolism inEscherichia coli.J Bacteriol, 2005,187: 3171−3179.
[38] Tatarko M, Romeo T. Disruption of a global regulatory gene to enhance central carbon flux into phenylalanine biosynthesis inEscherichia coli.Curr Microbiol, 2001,43: 26−32.
[39] Wahl A, El Massaoudi M, Schipper D,et al. Serial13C-based flux analysis of an L-phenylalanine-producingE. colistrain using the sensor reactor.Biotech Prog, 2004,20: 706−714.
[40] Okuda S, Yamada T, Hamajima M,et al. KEGG Atlas mapping for global analysis of metabolic pathways.Nucleic Acids Res, 2008, 36: W423−W426.
[41] Jensen LJ, Saric J, Bork P. Literature mining for the biologist: from information retrieval to biological discovery.Nat Rev Gene, 2006, 7: 119−129.
[42] Ananiadou S, Kell DB, Tsujii J. Text mining and its potential applications in systems biology.Trends in Biotech, 2006, 24: 571−579.
[43] Leitner F, Valencia A. A text-mining perspective on the requirements for electronically annotated abstracts.FEBS Lett, 2008, 582: 1178−1181.
[44] Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction.Nat Protoc, 2010, 5: 93−121.
Reconstruction and application of genome-scale metabolic network model
Liming Liu, and Jian Chen
State Key Laboratory of Food Science and Technology,Jiangnan University,Wuxi214122,China
Received:May 9, 2010;Accepted:August 3, 2010
Supported by:National Outstanding Youth Foundation of China (No. 20625619), Foundation for the Author of National Excellent Doctoral Dissertation of China (No. 200962).
Corresponding author:Liming Liu. Tel: +86-510-85918307; E-mail: mingll@jiangnan.edu.cn
Jian Chen. E-mail: jchen@jiangnan.edu.cn
国家杰出青年基金 (No. 20625619),全国优秀博士学位论文作者专项资金 (No. 200962) 资助。
猜你喜欢
肝博士(2022年3期)2022-06-30
海外星云(2021年9期)2021-10-14
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
少儿美术(2019年1期)2019-12-14
小哥白尼(趣味科学)(2018年6期)2018-09-14
中成药(2018年7期)2018-08-04
中学生数理化·高一版(2018年6期)2018-07-09
中国体育教练员(2017年3期)2018-01-19
