
单环刺螠线粒体基因组全序列的获得
——长PCR结合鸟枪法测序
2010-09-24 08:09吴志刚
海洋科学 2010年12期
申 欣, 吴志刚,3
(1. 淮海工学院 海洋学院, 江苏 连云港 222005; 2. 中国科学院 海洋研究所, 山东 青岛 266071; 3. 加州大学河滨分校, 美国 加利福尼亚州 92521)
单环刺螠线粒体基因组全序列的获得
——长PCR结合鸟枪法测序
申 欣1,2, 吴志刚2,3
(1. 淮海工学院 海洋学院, 江苏 连云港 222005; 2. 中国科学院 海洋研究所, 山东 青岛 266071; 3. 加州大学河滨分校, 美国 加利福尼亚州 92521)
由于具有单基因所不可比拟的优势, 线粒体基因组现已成为后生动物种群遗传和分子系统发育研究中一个重要的信息来源。本研究选取螠虫动物的代表物种——单环刺螠(Urechis unicinctus), 详细阐述了利用长PCR方法扩增其线粒体DNA, 获得约15 kb的扩增产物, 进而构建shotgun文库, 最终成功获得单环刺螠线粒体基因组全序列的流程。本实验流程样品需求量少、设备要求低、简便快捷。
线粒体基因组; 长PCR; 鸟枪法; 单环刺螠(Urechis unicinctus)
由于分子生物学和基因组学的快速发展, 科学家们逐渐认识到生命形式起源和进化的密码蕴涵在DNA链中, 核苷酸的组合变化和基因的排列顺序提供了有价值的系统发育信息。与单个基因相比, 线粒体基因组是一个完整的体系, 具有信息量丰富(包括RNA二级结构和基因排列顺序等)和系统发育树结构稳定等优点[1~3]。由于线粒体基因组具有诸多优势,使其成为研究后生动物关键类群的起源、进化、系统发育关系及群体遗传分化的理想材料[4~6]。
目前可以通过 3种方法获得后生动物线粒体基因组: 常规PCR方法、物理分离方法和长PCR方法。常规 PCR方法操作简单、对样品需求量少, 但容易受到线粒体假基因和串联重复区的干扰; 而物理方法分离线粒体基因组 DNA, 则存在对仪器要求高、样品需求量大等缺点[2], 这在一定程度上制约着线粒体基因组研究的快速发展。本研究选取 虫动物代表物种——单环刺螠(Urechis unicintus), 利用长PCR方法扩增其线粒体DNA进而对长PCR产物构建shotgun文库, 最终成功获得单环刺螠线粒体基因组全序列。本实验流程的建立为测定其他海洋无脊椎动物的线粒体基因组提供较高的参考价值。
1 材料与方法
1.1 材料
鲜活的单环刺螠样品购于水产品市场, 体长约15 cm, 呈长囊形, 身体呈淡红色, 由吻及躯干两部分组成。
1.2 方法
1.2.1 单环刺螠线粒体分离及线粒体DNA提取
取1g单环刺螠肌肉组织, 在洁净培养皿里将其剪碎。将剪碎的肌肉组织转入匀浆管里进一步匀浆。匀浆时, 按照比例[匀浆缓冲液体积(mL) : 组织质量(g) =2 : 1]加入匀浆缓冲液(100 mmol/L Tris-HCl, 250 mmol/L蔗糖, 10 mmol/L EDTA, pH 7.4); 匀浆10次左右至无块状组织, 把匀浆液转移至 1.5 mL 无菌Eppendorf管中; 4 ℃、2 300 g离心10 min, 弃沉淀,转上清于新的无菌Eppendorf管中; 重复上述步骤1次; 取上清液于4 ℃、20 000 g离心20 min沉淀线粒体, 弃上清。然后, 加750 µL裂解液(100 mmol/L Tris-HCl, 100 mmol/L EDTA, 1.5 mol/L NaCl, 1%CTAB, pH 8.0)裂解提取DNA, 所得DNA作为常规PCR和长PCR的模板。
1.2.2 单环刺螠线粒体DNA的PCR扩增
利用后生动物cox1基因的通用引物(LCO: GGT CAA CAA ATC ATA AAG ATA TTG G, HCO: TAA ACT TCA GGG TGA CCA AAA AAT CA)[7]采取常规PCR的扩增方案获得cox1基因的部分片段, 测序后得到单环刺螠cox1基因的部分序列, 然后根据此序列设计长PCR引物(cox1F: TCA CCC TGT TCC TAC TCC TCC ACC AA,cox1R: CCT ACG ACC AGA ACG AAT GCC TTT AT), 扩增余下的线粒体基因组区域, 从而获得单环刺螠线粒体DNA(图1)。长PCR扩增的反应体系为: 18 µL ddH2O、2.5 µL LA-Buffer(Mg2+plus, Takara)、0.5 µL dNTP(分别为 10 mmol/L)、1 µLcox1F(5 μmol/L)、1 µLcox1R(5 μmol/L)、1 µL LA Taq聚合酶(1 unit, Takara)和1µL模板。长PCR扩增的反应条件为: 94 ℃变性2 min, 然后34个循环(94 ℃20 s, 54 ℃退火45 s, 65 ℃延伸13 min), 最后72 ℃延伸10 min, 最终获得了大小约15 kb的线粒体DNA片段(图2)。
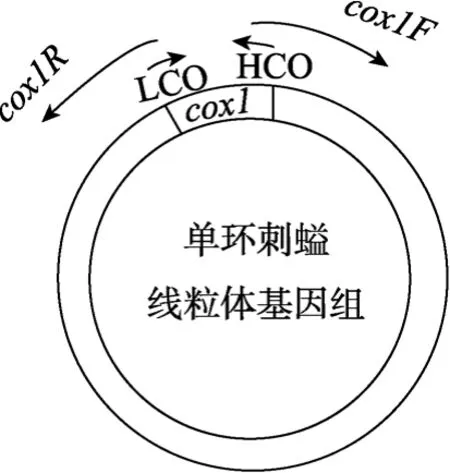
图1 单环刺螠线粒体基因组的扩增方法Fig. 1 The amplification strategy ofU. unicinctusmitochondrial genome
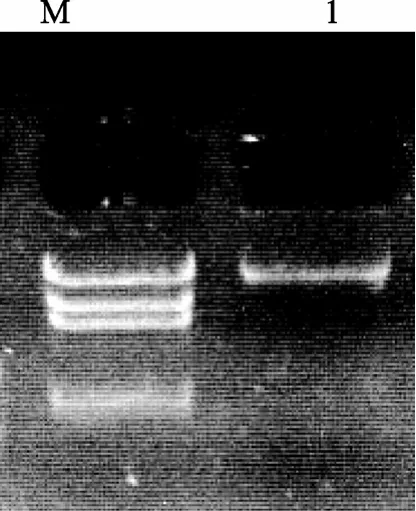
图2 单环刺螠线粒体DNA长PCR扩增产物Fig. 2 Long-PCR amplification product ofU. unicinctusmitochondrial DNA
1.2.3 单环刺螠线粒体DNA随机破碎及shotgun文库的构建
把长度约为15 kb的PCR产物置于冰上用超声破碎仪破碎(200 V, 持续时间0.2 s, 破碎3~5次), 在1%琼脂糖凝胶上检测线粒体 DNA的破碎情况。在进行末端修复后, 利用胶回收试剂盒(QIAGEN Gel Purification Kit)回收破碎后的 PCR产物, 纯化后的产物随后与pUC18(Takara)平端载体连接16 h。连接产物于透析膜上透析1 h。取1 µL透析后连接产物用BIO-RAD电转仪电击(电压: 2.1 kV)转入49 µL大肠杆菌(Escherichia coli)DH10B, 电击产物迅速转入1 mL SOC培养基中, 180 r/min复苏1 h, 取100 µL菌液在LB平板(含氨苄青霉素100b mg/L、X-gal 40 mg/L和IPTG 24 mg/L)上过夜培养。
1.2.4 重组质粒测序
根据pUC18载体自身序列设计的M13+和M13−引物在ABI 3730 x1自动测序仪上对阳性克隆进行测序。单环刺螠线粒体基因组大部分序列是通过克隆测序获得; 在克隆没有覆盖到的区域以及低质量值的位置, 通过设计引物进行常规 PCR扩增, 然后对常规PCR产物进行测序获得峰图。
1.2.5 序列拼接及基因注释
将测序峰图文件通过Phred-phrap软件进行序列组装[8,9], 然后通过Consed软件对序列拼接的精确性和测序质量进行检查[10], 从而获得单环刺螠线粒体基因组全序列。蛋白质编码基因和核糖体RNA基因通过DOGMA软件进行注释[11], 而转运RNA基因通过tRNAscan-SE 1.21软件进行预测[12]。
2 结果与讨论
2.1 单环刺螠线粒体基因组结构及组成
单环刺螠线粒体基因组全长为15 761 bp, 与其他大多数后生动物线粒体基因组的基因组成相同,共编码37个基因, 其中包括13个蛋白质编码基因、22个转运RNA基因和两个核糖体RNA基因。基因排列顺序与美洲刺螠(Urechis caupo)线粒体基因组完全相同[13]。单环刺螠、美洲刺螠与已知的环节动物线粒体基因组在基因分布上具有一个共同的特征:所有基因都在同一条链上编码[14~17]。
2.2 单环刺螠线粒体基因组的非编码区
单环刺螠线粒体基因组与已测定的其他后生动物线粒体基因组一样, 包含了几个非编码区。非编码区总长 997 bp, 其中最长的非编码区长度为923 bp, 余下的74bp分布在线粒体环上其他的 15个区域, 除了在tRNASer(AGN)和cox3之间的非编码区域长达42 bp之外, 其余非编码区的长度均在1~5 bp。单环刺螠、美洲刺螠与环节动物代表物种的最大非编码区的长度和 AT含量见表1。
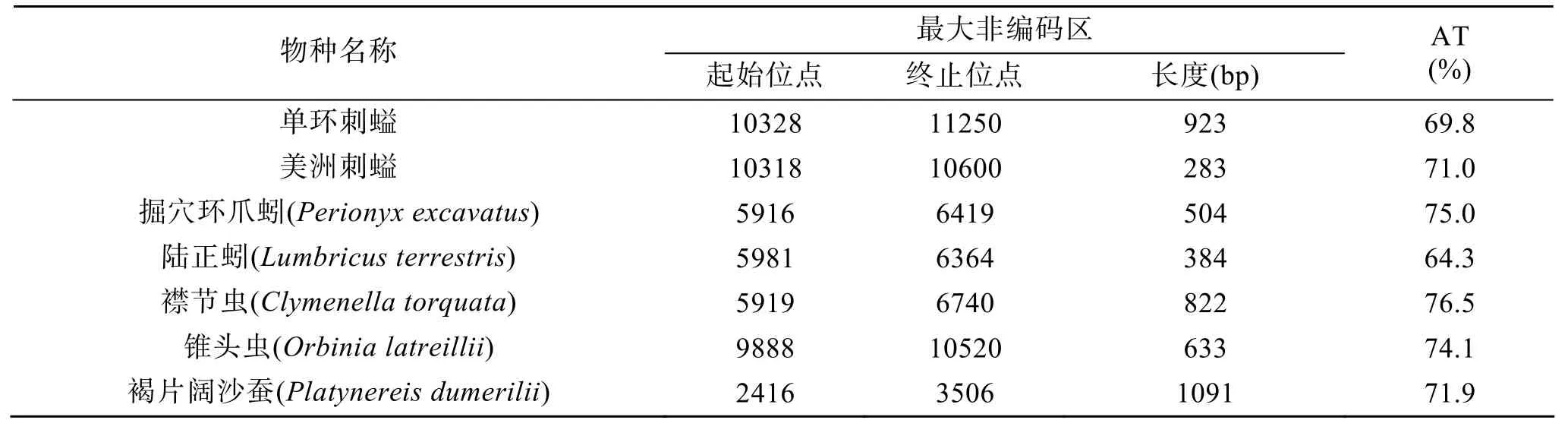
表1 单环刺螠、美洲刺螠与环节动物代表物种线粒体基因组的最大非编码区Tab. 1 The largest non-coding region of available echiurans and annelids mitochondrial genomes
2.3 基因排列顺序Boore等[18]认为基因排列的比较可为系统发育研究提供一些有价值的信息, 特别是在探讨一些古老的进化关系时。比较螠虫动物和环节动物的线粒体基因组的基因排列, 呈现出较高的保守性。环节动物门内的基因顺序相对保守[14], 单环刺螠线粒体基因组有 3个基因区块与环节动物具有相同的基因排列顺序, 分别是trnT-nad4L-nad4、nad1-trnI和trnQ-
nad6-cob-trnW-atp6-trnR-trnH-nad5-trnF(图 3)。这些保守的基因排列顺序在线粒体基因组层次上支持虫动物和环节动物具有亲缘关系。
2.4 3种线粒体基因组获取方法的比较
目前可通过 3种方法获得后生动物线粒体基因组: 常规PCR方法、物理分离方法和长PCR方法。常规PCR的方法根据后生动物线粒体基因组的通用引物, 或通过近源物种的线粒体基因组全序列, 设计出覆盖全基因组的引物, 采用常规 PCR技术对线粒体基因组进行扩增。常规PCR的方法具有操作简单、对样品需求量少等优点, 但在操作中容易受到线粒体假基因和串联重复区的干扰[2,19]。而基于物理方法分离线粒体基因组 DNA(主要包括密度梯度离心法和差速离心法等), 则存在对仪器要求高、样品需求量大等缺点。基于长PCR的方法对模板质量、引物特异性和聚合酶都有相对较为严格的要求[2]。本研究以1g单环刺螠肌肉组织为实验材料, 利用长PCR方法扩增其线粒体 DNA, 获得约 15kb的扩增产物,进而超声随机破碎、构建shotgun文库, 最终成功获得单环刺螠线粒体基因组全序列的流程。
3 结论

图3 螠虫动物和环节动物线粒体基因组基因排列的比较Fig. 3 Gene order of mitochondrial genomes from Echiura and Annelida
本研究通过长 PCR方法, 高效、快捷地获得高纯度的单环刺螠线粒体 DNA, 通过构建 shotgun文库顺利获得了单环刺螠线粒体基因组全序列, 通过生物信息学软件进行了序列组装和基因预测。结果显示, 单环刺螠线粒体基因组 DNA为双链环状DNA, 全长15 761 bp; 与后生动物线粒体基因组典型的基因组成相同, 包含37个基因, 其中13个蛋白质编码基因、22个tRNA基因和2个核糖体RNA基因。本实验方法与物理分离方法相比具有样品需求量少、设备要求低和简便快捷等优点, 而且同时避免了常规PCR容易受到假基因和串联重复区干扰等问题, 为测定其他海洋无脊椎动物的线粒体基因组提供参考。
[1] Boore J L, Collins T M, Stanton D,et al.Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements[J].Nature, 1995, 376: 163-165.
[2] 尚娜, 周志军.动物线粒体基因组全序列测定的研究策略[J].河池学院学报, 2008, 28(2): 62-65.
[3] Lavrov D V, Lang B F.Poriferan mtDNA and animal phylogeny based on mitochondrial gene arrangements[J].Syst Biol, 2005, 54: 651-659.
[4] Helfenbein K G, Fourcade H M, Vanjani R G,et al.The mitochondrial genome ofParaspadella gotoiis highly reduced and reveals that chaetognaths are a sister group to protostomes[J].Proc Natl Acad Sci USA, 2004, 101:10 639-10 643.
[5] Dellaporta S L, Xu A, Sagasser S,et al.Mitochondrial genome ofTrichoplax adhaerenssupports placozoa as the basal lower metazoan phylum[J].Proc Natl Acad Sci USA, 2006, 103: 8 751-8 756.
[6] Shen X, Ma X Y, Ren J F,et al.A close phylogenetic relationship between Sipuncula and Annelida evidenced from the complete mitochondrial genome sequence ofPhascolosoma esculenta[J].BMC Genomics, 2009, 10:136-146.
[7] Folmer O, Black M, Hoeh W,et al.DNA primers for amplification of mitochondrial cytochrome coxidase subunit I from diverse metazoan invertebrates[J].Mol/L Mar Biol Biotechnol, 1994, 3(5): 294-299.
[8] Ewing B, Hillier L, Wendl M C,et al.Base-calling of
automated sequencer traces using phred. I. Accuracy assessment[J].Genome Res, 1998, 8(3): 175-185.
[9] Ewing B, Green P.Base-calling of automated sequencer traces using Phred. II. Error probabilities[J].Genome Res, 1998, 8(3): 186-194.
[10] Gordon D, Abajian C, Green P.Consed: A graphical tool for sequence finishing[J].Genome Res, 1998, 8(3):195-202.
[11] Wyman S K, Jansen R K, Boore J L.Automatic annotation of organellar genomes with DOGMA[J]. Bioinformatics, 2004, 20(17): 3 252-3 255.
[12] Lowe T M, Eddy S R.tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence[J].Nucleic Acids Res, 1997, 25: 955-964.
[13] Boore J L.Complete mitochondrial genome sequence ofUrechis caupo, a representative of the phylum Echiura[J].BMC Genomics, 2004, 5(1): 67-74.
[14] Jennings R M, Halanych K M.Mitochondrial genomes ofClymenella torquata(Maldanidae) andRiftia pachyptila(Siboglinidae): evidence for conserved gene order in annelida[J].Mol/L Biol Evol, 2005, 22(2):210-222.
[15] Bleidorn C, Podsiadlowski L, Bartolomaeus T.The complete mitochondrial genome of the orbiniid polychaeteOrbinia latreillii(Annelida, Orbiniidae)—A novel gene order for Annelida and implications for annelid phylogeny[J].Gene, 2006, 370: 96-103.
[16] Boore J L, Brown W M.Mitochondrial genomes ofGalathealinum,Helobdella, andPlatynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa[J].Mol/L Biol Evol, 2000,17(1): 87-106.
[17] Boore J L, Brown W M.Complete sequence of the mitochondrial DNA of the annelid wormLumbricus terrestris[J].Genetics, 1995, 141(1): 305-319.
[18] Boore J L, Brown W M.Big trees from little genomes:mitochondrial gene order as a phylogenetic tool[J].Curr Opin Genet Dev, 1998, 8: 668-674.
[19] Ren J F, Liu X, Zhang G F,et al.“Tandem duplication-random loss” is not a real feature of oyster mitochondrial genomes[J].BMC Genomics, 2009, 19(10):84-89.
Received: Mar., 5, 2010
Key words:mitochondrial genome; long PCR; shotgun;Urechis unicintus
Abstract:Due to their many advantages over single genes, mitochondrial genomes have become an important source of information in metazoan population genetics and molecular phylogenetic studies. In this study,Urechis unicintus, a represent species of Echiura, was chosen to be as an example to clearly elaborate the process of long PCR amplification of its mitochondrial DNA. The long PCR amplified product was about 15kb. Then the shotgun library was constructed and the entire mitochondrial genome sequence was successfully obtained. The results show that the long PCR-based method is less demand of devices and amounts of tissue compared with physical separation method.
(本文编辑:谭雪静)
Complete mitochondrial genome sequence of Urechis unicinctus accomplished by long PCR combined with shotgun sequencing
SHEN Xin1,2, WU Zhi-gang2,3
(1. College of Marine Science, Huaihai Institute of Technology, Lianyungang 222005, China; 2. Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China; 3. University of California, Riverside, CA 92521, USA)
Q959.195
A
1000-3096(2010)12-0026-04
2010-03-05;
2010-08-04
江苏省自然科学基金项目(BK2007066); 江苏省海洋生物技术重点建设实验室课题(2009HS13)
申欣(1981-), 男, 山东成武人, 讲师, 博士, 主要从事线粒体基因组学的研究, E-mail: shenthin@163.com
猜你喜欢
食品研究与开发(2022年1期)2022-01-24
水产学杂志(2021年4期)2021-10-18
今日农业(2021年11期)2021-08-13
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中西医结合肝病杂志(2020年2期)2020-10-27
中成药(2018年7期)2018-08-04
公民与法治(2016年24期)2016-05-17
癌变·畸变·突变(2014年1期)2014-03-01
遗传(2014年3期)2014-02-28
