
醋酸铑催化有机反应研究进展
2010-09-08 02:21:50钱明诚许孝良李小年
化工生产与技术 2010年6期
王 红 钱明诚 许孝良 叶 宁 马 磊 李小年
(浙江工业大学 化学工程与材料学院,杭州 310032)
醋酸铑催化有机反应研究进展
王 红 钱明诚 许孝良 叶 宁 马 磊 李小年
(浙江工业大学 化学工程与材料学院,杭州 310032)
叙述了醋酸铑催化分解α-重氮羰基化合物生成的金属卡宾参与的有机反应,反应类型大体可以分为环化反应、插入反应和生成叶立德反应3类。铑(II)卡宾以及铑(II)金属卡宾与反应体系中杂原子作用生成的叶立德活性中间体,随后可以进行多种有机转化,作为一步合成多化学键、多官能团有机化合物的新方法,在天然产物和大分子化合物的合成中已得到了广泛的应用。
醋酸铑;金属卡宾;叶立德;有机反应
在早期的金属卡宾形成过程中,几乎所有的反应都是用Cu做催化剂完成的[4-5]。但由于Cu催化剂普遍具有很高的活性,以至于它产生的金属卡宾活性高而选择性比较低,使其反应过程变得比较复杂,应用范围受到一定的限制。除了Cu以外,化学家一直在寻找其他能够分解重氮化合物的过渡金属。1937年,Teyssie发现了第1个金属铑催化剂——醋酸铑(II)可以有效的催化重氮化合物的分解,至此金属卡宾化学进入了一个高速发展的新纪元,铑成为最有效并且最具普遍适应性的金属催化剂[6]。
不同于金属铜催化剂,2价铑具有其独特的结构。以最常用的2价金属铑催化剂Rh2(OAc)4为例:醋酸铑是以二聚体的形式存在的,4个醋酸根形成桥键与2个铑原子配合,而2个铑原子之间是单键,空间上为D4对称的八面体,每个铑原子在对称轴方向都有一个空的配位位置。Nakamura等通过计算认为在金属铑卡宾的形成过程中,2个铑原子中只有1个铑原子与重氮碳配位,另1个铑原子通过接受电子提高相应卡宾的亲电性以有利于Rh—C键的断裂从而完成分解反应[7]。正是这些结构上的特点,使得醋酸铑的反应条件温和、化学选择性比较高,从而使其成为使用最为广泛的非手性催化剂。
自从上世纪70年代,Teyssie研究小组报道了二聚醋酸铑 Rh2(CH3CO2)4催化的以 α-重氮羰基化合物为原料的一系列反应具有很高的化学、区域和立体选择性以来,Rh2(CH3CO2)4作为一种重要的 有机金属催化剂,在有机合成领域扮演着越来越重要的角色,Rh2(CH3CO2)4催化的有机反应也越来越多。醋酸铑催化分解α-重氮羰基化合物生成的金属卡宾的反应类型大体可以分为3类:环化反应、插入反应和生成叶立德的反应,这些反应在有机合成领域已经获得了广泛的应用[8]。
1 催化金属卡宾插入反应
1.1 C—H键
醋酸铑催化的分子内的C—H键的插入反应已有很多报道并在有机合成中得到广泛的应用[9]。其中,铑(II)金属卡宾分子内的1,5 C—H键的插入反应由于反应过程中生成稳定的六元环过渡态而最为常见。
受电子效应、空间效应和与铑(II)金属卡宾相邻取代基的影响,铑(II)金属卡宾也会发生分子内1,3,1,4,1,6或1,7 C—H键的插入反应。如Shi课题组报道了醋酸铑催化β-对甲苯酰基-α-偶氮化合物,生成的金属卡宾接着发生1,3 C—H键的插入反应,以中等产率得到了一系列环丙烷的衍生物[10];Nanada等的研究表明当C—H键被邻位的氧原子或氮原子等杂原子活化时,铑(II)金属卡宾会发生分子内的1,4和1,7 C—H键的插入反应生成相应的环状化合物[11]。Doyle等也利用铑(II)金属卡宾的C—H键的插入反应成功合成了14元环的大环内酯物[12]。
1982年Taber以长链α-偶氮-β-酮酸酯为原料,醋酸铑为催化剂,在室温下生成的金属卡宾发生分子内的C—H键的插入反应合成了一系列的环戊二烯衍生物[13]。这一合成五元环化合物的方法一经报道立刻引起了人们很大的兴趣。Douglass等人后来对此类反应又进行了深入的研究,研究表明,此类反应不仅有较好的产率,而且C—H的插入也具有很好的立体专一性,如果反应的C—H中的碳原子连有3个不同的原子或基团,插入环化后会得到1个含有手性碳原子的光学活性单一的五元环酮,这为合成具有光学活性的五元环化合物提供了一个简便可行的方法[14]。反应式为(Me为甲基):
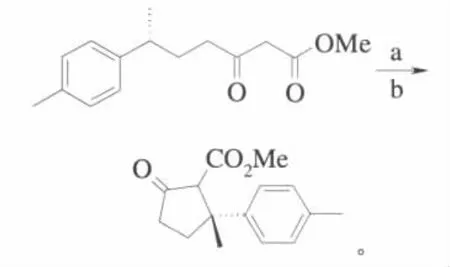
其中,a:对甲苯磺酰叠氮、三乙基胺;b:Rh2(OAc)4、CH2Cl2。
1.4.1 近期疗效[6] 治疗后,按照WHO标准对2组患者近期疗效进行评价,疗效等级分为完全缓解(CR)、部分缓解(PR)、稳定(SD)及进展(PD),总有效率=(CR+PR)/总数×100%。
与氧原子或氮原子相邻的C—H键,很容易与金属卡宾发生C—H键的插入反应。但是和硫原子相邻的C—H键,却很难和金属卡宾发生类似的分子内的插入反应。对反应机理的研究表明,在此类反应过程中形成的金属卡宾会优先被底物中的硫原子捕获生成相应的硫叶立德,从而抑制了分子内C—H键的插入反应。选择合适的反应底物和催化剂,有效阻止反应过程中硫叶立德的形成,一直是化学家研究的热点[15]。
2004年,Paul首次报道了醋酸铑催化偶氮内酯生成相应的金属卡宾,接着和与硫原子相邻的C—H键发生分子内的1,5-插入反应,以51%~84%的产率得到了一系列双桥氢化噻吩呋喃酮。反应过程中没
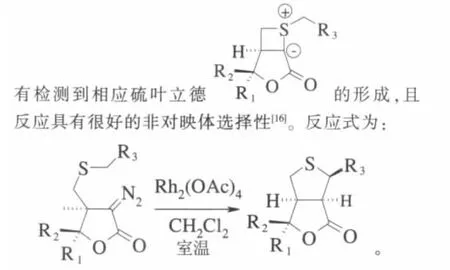
其中,R1=苯基、甲基、异丙基,R2=H、甲基,R3=苄基、异丙基、丙基。
1.2 N—H键
自从1974年,首次报道了醋酸铑催化金属卡宾的N—H键的插入反应以来,此领域的研究工作引起了人们的广泛关注[17]。醋酸铑催化的金属卡宾分子内N—H键的插入反应提供了一种温和且高效合成4-、5-和6-元氮杂环化合物的方法。如在工业化生产双环β-内酰胺沙纳霉素的过程中,铑(II)催化的分子内N—H键的插入反应是最为关键的一步[18]。但由于醋酸铑催化的金属卡宾的分子内N—H键的插入反应和分子内C—H键的插入反应存在竞争,使得此类反应往往得不到单一的产物。Papoport曾报道在醋酸铑催化的金属卡宾的分子内N—H键的插入反应合成3-氧化哌啶的过程中,同样分离得到了C—H键的插入反应的产物五元碳环化合物[19]。因而,利用醋酸铑催化相应金属卡宾分子内的N—H键的插入反应来合成一些复杂结构化合物的方法受到一定限制。
在温和的反应条件下,过渡金属配合物可以催化α-重氮化合物与胺的分子间N—H键的插入反应,这是合成氨基酸的一种有意义的方法。其中,醋酸铑催化的金属卡宾与胺的分子间N—H键的插入反应被广泛用来合成许多天然产物,也是合成药物中间体常用的方法。如Paulissen、Osipov、Haigh和Moody先后分别报道了醋酸铑催化重氮乙酸酯、α-重氮三氟丙酸乙酯、α-重氮苯乙酸乙酯等与胺分子当中N—H键发生分子间的插入反应,合成了一系列 α-氨基酸[20-22]。
黄丹课题组在以上研究的基础上,以廉价的芳基乙酸酯为起始原料,经重氮化后,在温和的反应条件下,用醋酸铑催化α-重氮芳基化合物与酰胺的分子间的N—H键插入反应,以中等产率合成得到芳基甘氨酸的衍生物N-乙酰基芳基甘氨酸甲酯,该产物可以用作金属配体,手性和非手性的非天然氨基酸的合成,具有广泛的应用前景[23]。
2 催化金属卡宾的环化反应
将过渡金属催化剂应用于环加成是金属有机化学研究的热点。许多难以发生或产率很低的环化反应在过渡金属催化剂的作用下得到了改善。铑(II)催化剂催化的环加成反应由于具有高对映选择性、反应时间短、转化率高等优点而引起人们的广泛关注。醋酸铑应用于环丙烷化反应可合成最简单的三元环,通过金属卡宾与不饱和双键的加成反应可以得到许多重要的环丙烷衍生物。在这种分子间的环化反应中,产物的立体选择性一直是人们关注的热点。Willian报道了以醋酸铑为催化剂2-偶氮茚酮和取代烯烃的分子间的环化反应[24]:
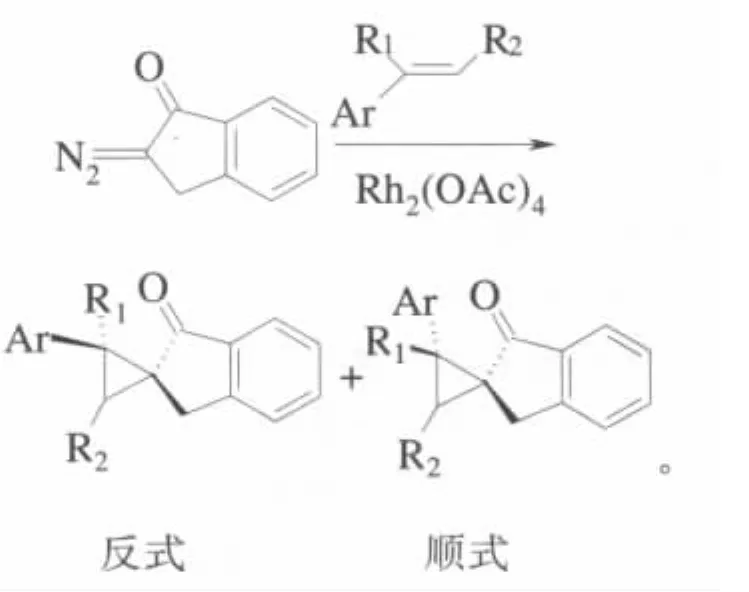
其中Ar为芳香基。研究表明,醋酸铑可以有效地催化2-偶氮茚酮和取代烯烃的环丙烷化反应,以中等产率得到了一系列的取代环丙烷衍生物。取代烯烃的结构不仅影响了反应的最终产率,而且还和产物中反式异构体和顺式异构体的比例有关。产物具有较好的对映选择性,顺式和反式异构体的比例主要取决于过渡态金属卡宾和烯烃形成的∏-络合物的空间取向。
随后,Ghanem研究小组在合成4-苯甲酸-2-苯基-1-三氟甲基环丙烷基甲基醚中也成功地利用醋酸铑催化重氮化合物和苯乙烯的环丙烷化反应,合成了相应的环丙烷衍生物,并通过对产物的分离,得到了顺式和反式异构体[25]。
氮杂环丙烷能够进行区域性选择的立体选择开环,是很有价值的有机合成中间体,许多生物活性物质中也都含有氮杂环丙烷的结构单元,因而其合成方法的研究引起了人们的广泛兴趣。利用重氮化合物直接对亚胺加成来合成氮杂环丙烷的研究一直是此领域的研究热点[26]。此合成方法受到重氮化合物的结构和催化剂活性的制约,选择合适的反应底物和高活性的催化剂是反应成功的关键。
李志成等报道了利用醋酸铑催化重氮苄基膦酸酯与芳香亚胺的氮杂环丙烷,高选择性地、立体专一地以中等产率得到了氮杂环丙烷-2-膦酸酯[27]:
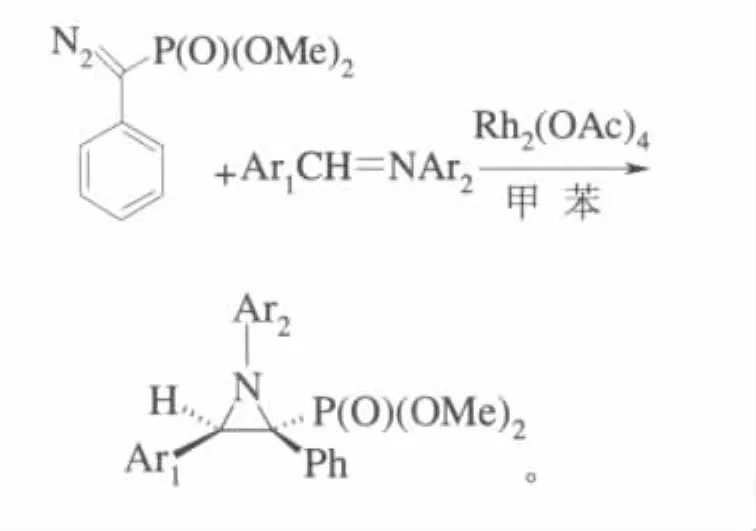
3 催化叶立德反应
金属催化剂催化分解α-重氮羰基化合物生成的金属卡宾除了发生插入、环化反应外,还有一种是生成叶立德的反应。相对于已经广泛应用于有机合成中的环化和插入反应,催化分解的叶立德在有机反应中的应用直到近年才为人们所认识。叶立德可以被看作是一种特别的离子,通过碳负离子与其相邻的带正电的原子(氧、碘、硫和氮等)组成。作为一种重要的有机合成手段,形成的叶立德主要有3种反应:2,3-Sigmatropic重排、Stevens重排和偶极环加成[28]。
3.1 2,3-Sigmatropic重排
含有烯丙基的叶立德的2,3-Sigmatropic重排反应是有机化学中化学键重组的重要反应之一,研究的比较早而且深入的是烯丙基硫叶立德的反应[29]。David报道了醋酸铑催化α-重氮化合物生成的金属卡宾,与一系列的烯丙基硫醚生成相应的硫叶立德,接着发生2,3-Sigmatropic重排的反应:

其中,R=甲基、苯基,X=甲酸乙酯、三甲基硅。他们的研究结果表明,当以三甲基硅重氮甲烷为反应底物时,2,3-Sigmatropic重排的反应会得到更高的收率[30]。
近来,烯丙基氮和氧叶立德的2,3-Sigmatropic重排反应也有了报道和研究。这类反应作为关键的合成步骤,在具有多官能团的复杂的天然产物分子的合成中得到应用[31]。例如在生物碱manzamine A的全合成中,Clark利用烯丙基氮叶立德的2,3-Sigmatropic重排反应,成功实现了复杂的manzamine A分子中CE环系的构建[32]。
3.2 Stevens重排
醋酸铑催化的分子内和分子间的硫,氧和氮叶立德的[1,2]-Shift重排反应也是常见的叶立德重排反应。此类反应具有较高的立体选择性,是一种有效的合成含氧、氮杂环有机分子的常用方法[33]。如在天然产物(-)-Epilupinine合成过程中涉及到的环状氮叶立德的Stevens[1,2]-Shift反应是合成目标产物中关键的一步[34]。West等也曾报道了以醋酸铑催化α-重氮羰基化合物,生成环状氧叶立德,然后利用氧叶立德的[1,2]-Shift反应合成相应的环醚的方法[35]:
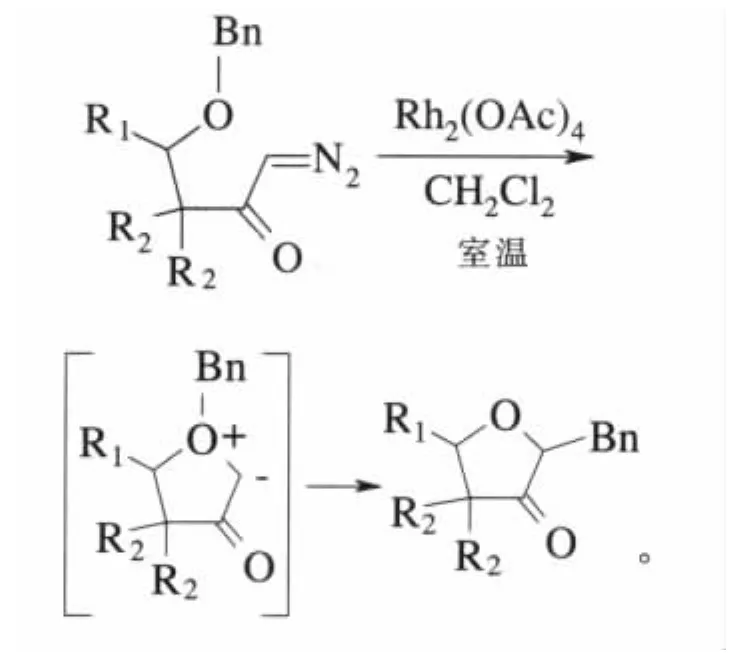
3.3 偶极环加成
醋酸铑催化生成的金属卡宾生成叶立德的反应,早期的工作主要集中于2,3-Sigmatropic、Stevens重排反应的研究。近年来,铑(II)金属卡宾与醛酮、亚胺作用生成的分子内或分子间的羰基叶立德和亚胺叶立德中间体的高反应活性逐渐引起了人们的注意。这类叶立德的主要反应是与亲二烯体发生偶极环加成从而构建多取代的氧、氮杂环等。这些化合物往往是合成许多活性天然产物的重要的结构单元,这一方面的研究工作已经渐渐引起了人们的研究兴趣,已有大量的文献报道[36]。
醋酸铑催化的重氮化合物与醛酮的分子间反应过程比较复杂,反应有很多途径。既可单独发生羰基叶立德中间体的自身环化反应或与亲偶极试剂结合的1,3-偶极环加成反应,又有可能2种反应同时进行,此外还涉及到产物的立体选择性。目前研究比较多的主要是1,3-偶极环加成反应[37]。最早的α-重氮羰基化合物与醛的分子间反应的研究,是Huisgen报道的重氮丙二酸二甲酯在醋酸铑的催化下与苯甲醛的反应。金属卡宾与2分子的醛作用生成二氧戊环,但反应的选择性比较差[38]:
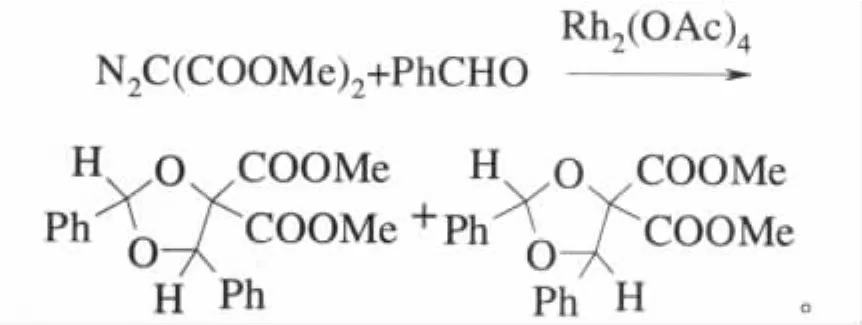
复杂反应历程的控制是非常令人感兴趣的。随后,Doyle等以醋酸铑催化重氮乙酸酯和芳香醛地反应为例,对一系列分子间羰基叶立德的形成以及环加成的立体控制过程进行了研究。研究结果表明,不同的芳香醛底物和催化剂的选择对最终的反应产物的立体选择性有很大的影响,这可能与中间体羰基叶立德的形成和稳定性有很大的关系[39]。
4 结语
醋酸铑催化分解α-重氮化合物生成的金属卡宾以及金属卡宾与体系中杂原子作用生成的叶立德活性中间体,可以进行非常多的有机转化,作为一步合成多化学键、多官能团有机化合物的新方法,已经得到广泛的应用并且发表了很多相关的研究报告。但在这个领域仍然存在挑战,如α-重氮化合物和反应底物的结构对形成的金属卡宾和叶立德稳定性的影响以及各类反应机理的研究,选择合适的反应底物对各类反应的化学选择性和立体选择性的影响,新的反应体系的发现等等。这些方面的研究必将引起化学家的极大关注,醋酸铑作为一种常见的金属催化剂也有望得到新的发展。
[1]Witting G,Geissler G.Zur reaktionsweise des pentaphenylphosphors und einiger derivate[J].Justus Liebigs Ann Chem,1953,580:44-48.
[2]Padwa A,Hornbuckle S F.Ylide formation from the reaction of carbenes and carbenoids with heteroatom lone pairs[J].Chem Rev,1991,91:263-309.
[3]Doyle M P,Mckervey M A,YE T.In modern catalytic methods for organic synthesis with diazo compounds[M].New York:John Wiley&sons,Inc,1997.
[4]Nozaki H,Moriuti S,Yamabe M,et al.Reactions of diphenyldiazomethane in the presence of bis(acetylacetonato)copper (II). Modified diphenylmethylene reactions[J].Tetrahedron Lett,1966,7(1):59-63.
[5]Corey E J,Myersy A G.Efficient synthesis and intramolecular cyclopropanationofunsaturateddiazoaceticester[J].Tetrahedron Lett,1984,25(33):3559-3562.
[6]Paulissenen R,Reimlinger H,Hayer E,et al.Metal catalysed reactions of dizaocompounds-II Insertion in the hydroxylicbond[J].Tetrahedron Lett,1973,14:2233.
[7]Nakamura E,Yoshikai N,Yamamkana M.Mechanism of C—H bond activation/C—C bond formation reaction between diazo compound and alkane catalyzed by dirhodium tetracarboxylate[J].J Am Chem Soc,2002,124:7181-7192.
[8]Doyle M P,Mckervey M A,Ye T.Modern catalytic methods for organic synthesis with diazo compounds[M].New York:John Wiley&Son,1998.
[9]Davies H M L,Beckwith R E J.Catalytic enantioselective C—H activation by means of metal carbenoid-induced C—H insertion[J].Chem Rev,2003,103:2861-2904.
[10]Shi W F,Zhang B,Zhang J,et al.Stereoselective intramolecular 1,3 C—H insertion in Rh(II)carbene reaction[J].Org Lett,2005,7(14):3103-3106.
[11]Nanada M,Hashimoto S I.Enantioselective intramolecular C—H insertion route to a key intermediate for the synthesis of trinem antibiotics[J].Tetrahedron Lett,1998,39 (49):9063-9066.
[12]Doyle M P,Protopopova M N,Poulter C D,et al.Macrocyclic lactonesfrom dirhodium (II)-catalyzed in tramolecular cyclopropanation and carbon-hydrogen insertion[J].J Am Chem Soc,1995,117(27):7281-7282.
[13]Taber D F,Petty E H.General route to highly functionalized cyclopentane derivatives by intramolecular C—H insertion[J].J Org Chem,1982,47(24):4808-4809.
[14]Taber D F,Petty E H,Raman K.Enantioselective ring construction:synthesis of (+)-alpha.-cuparenone[J].J Am Chem Soc,1985,107:196-199.
[15]Harrowven D C,Helias N L,Moseley J D,et al.Medium ring synthesis by radical ipso-substitution[J].Chem Commun,2003,21:2658-2659.
[16]Skerryp S,Swain N A,Harrowven D C,et al.Intramolecular C—H insertions adjacent to sulfur for the diastereoselective synthesis of thienofuranones[J].Chem.Commun,2004,1772-1773.
[17]Paulissen R,Havez E,Hubert A J,et al.Transition metal catalysed reactions of diazocompounds part III a one-step synthesis of substituted furanes and esters[J].Tetrahedron Lett,1974,15(7):607-608.
[18]Reider P J,Grabowski E J J.Total synthesis of thienamycin:a new approach from aspartic acid[J].Tetrahedron lett,1982,23:2293-2296.
[19]Moyer M P,Feldman P L,Rapoport H.Intramolecular N—H,O—H,and S—H insertion reactions.synthesis of heterocycles from α-Diazo-β-keto esters[J].J Org Chem,1985,50:5233-5236.
[20]Paulissen R,Hayez E,Hubert A J,et al.Transition metal catalysed reactions of dizaocompounds-partⅢa one step synthesis of substituted furanes and ester[J].Tetrahedron lett,1974,25:607-608.
[21]Osipov S N,Sewald N,Kolomiets A F,et al.Synthesis of α-trifluoromethyl substituted α-amino acid derivatives from methyl 3,3,3-trifluoro-2-diazopropionate[J].Tetrahedron lett,1996,37:607-608.
[22]Alter E,Buck R T,Drysdale M J,et al.N—H insertion reactions of rhodium carbenoids part 1 preparation of amino acid and aminophosphonic acid derivatives[J].J Chem Soc,Perkin Trans 1,1996,24:2879-2884.
[23]姜国民,陈红霞,徐青箐,等.重氮化合物的N—H插入反应合成N-乙酰基-2-芳基甘氨酸酯[J].南通大学学报,2007,6(4):46-51.
[24]William B,John D,James B,et al.Stereoselectivity in the rhodium(II)acetate catalysed cyclopropanations of 2-diazo-1-indanone with styrenes[J].Tetrahedron Letter,2000,41(4):1491-1494.
[25]Ghanem A,Aboul-Enein H Y.On the solvent versatility in immobilized amylose tris (3,5-dimethylphenyl carbamate)Chiralstationary phasesin high-performance liquid chromatography: application to the asymmetic cyclopropanation of olefins[J].Analytic Chimica Acta,2005,548:26-32.
[26]Yang X,Zhang M,Hou X L,et.al.Sterecontrolled aziridination of imines via a sulfonium ylide route and mechanistic study[J].J Org Chem,2002,67:8097-8101.
[27]李志成,杜月华,胡文浩.氮杂环丙烷-2-磷酸酯的高立体选择性合成[J].广东化工,2007,8(34):13-15.
[28]Li A H,Dai L X,Aggarwal V K.Asymmetric ylide reactions:epoxidation,cyclopropanation,aziridination,olefination,and rearrangement[J].Chem Rev,1997,97(6):2341-2372.
[29]Kido F,Yamaji K,Sinha S C.Carbocyclic construction by the[2,3]sigmatropic rearrangement of cyclic sulfonium ylides.a new entry for the stereoselective synthesis of substituted cyclohexanones[J].Tetrahedron,1995,51 (28),7697-7714.
[30]Carter D S,Van Vranken D L.Metal-catalyzed ylide formation and[2,3]sigmatropic rearrangement of allyl sulfides with trimethylsilyldiazomethane[J].Tetrahedron Letter,1999,40(9):1617-1620.
[31]Pirrung M C,Brown W L,Rege S,et al.Total Synthesis of(+)-Griseofulvin[J].J Am Chem Soc,1991,113:8561-8562.
[32]Clark J S,Hodgson P B.An enantioselective synthesis of the CE ring system of the alkaloids manzamine A,E and F,and ircinal a[J].Tetrahedron Lett,1995,36(14):2519-2522.
[33]Ollisl W D,REY M,Sutherland I O.Base catalysed rearrangement involving ylide intermediate.Part 15.The mechanism of the stevens[1,2]rearrangement[J].J Chem Soc,Perkin Trans,1983,11:1009-1027.
[34]West F G,Naidu B N.Piperidines via ammonium yield[1,2]-shift:a concise,enantioselective route to (- )-Epilupinine from proline ester[J].J Am Chem Soc,1994,116:8420-8421.
[35]Eberlein T H,West G,Tester R W.The stevens[1,2]-shift of oxonium ylides:a route to substituted tetrahydrofuranones[J].J Org Chem,1992,57(12):3479-3482.
[36]Padwa A,Krumpe K E.Application of intramolecular carbenoid reaction in organic synthesis[J].Tetrahedron,1992,48(26):5385-5453.
[37]Mehta G,Muthusamy S.Tandem cyclization-cycloaddition reaction of rhodium generated carbenoids from α-diazo carbonyl compounds[J].Tetrahedron,2002,58(47):9477-9504.
[38]March P,Huisgen R.Carbonyl ylides from aldehydes and carbenes[J].J Am Chem Soc,1982,104(18):4952-4956.
[39]Doyle M P,Forbes D C,Protopopova M N.Stereocontrol in intermolecular dirhodium -catalyzed carbonyl ylides formation and reactions[J].J Org Chem,1997,62 (21):7210-7215.
The Development of Organic Reaction Catalysed by Rhodium Acetate
Wang Hong,Qian Mingcheng,Xu Xiaoliang,Ye Ning,Ma Lei,Li Xiaonian
(College of Chemical Engineering and Materials Science,Zhejiang University of Technology,Hangzhou 310032)
Transition-metal catalysts have been widely utilized in many organic syntheses.Rhodium acetate was found to be an efficient catalyst for organic reactions.The purpose of this review is to give an overview of the various reactions of the Rh2(OAc)4-stabilized carbenoids derived from α-dizao-oxo-compounds.The types of these reactions include cyclication reaction,insertion reaction and the formation of ylides.The results show that the Rh(Ⅱ)carbenoids and the reactive ylide intermediates produced from the Rh(Ⅱ)carbenoids with the heteroatom of the system can undergo many organic translations and give enormous new synthetic methods to prepare compounds containing many function groups in one step.
rhodium acetate;metal carbenoids;ylides;organic reactions
TQ032.4
A DOI10.3969/j.issn.1006-6829.2010.06.008
浙江省科技厅面上科研工业项目(2007C21125)
2010-09-30
猜你喜欢
中学生数理化·自主招生(2023年4期)2023-04-26 03:06:43
科学技术与工程(2020年34期)2021-01-08 05:43:32
石油石化绿色低碳(2019年6期)2019-01-14 01:16:16
设备管理与维修(2016年6期)2016-03-16 02:22:06
石油炼制与化工(2015年7期)2015-09-03 10:56:29
时尚北京(2015年1期)2015-01-30 00:00:35
石油炼制与化工(2014年9期)2014-04-06 19:35:03
食品工业科技(2014年13期)2014-03-11 18:17:06
无机化学学报(2014年6期)2014-02-28 17:31:59
无机化学学报(2014年1期)2014-02-28 17:30:01
