
过硫酸钾氧化——紫外光度法测定总氮方法的改进
2010-07-31 03:48邓泽华
中国新技术新产品 2010年19期
邓泽华
(安徽省蚌埠市环境监测站,安徽 蚌埠 233040)
序言:过硫酸钾氧化紫外光度法测定总氮方法存在的主要问题是:空白吸光度高;K2S2O8-NaOH碱性溶液不稳定,只能使用一周。针对上述问题,笔者做了大量试验,对测定总氮的方法提出了改进意见。
1 实验部分
1.1 仪器
①紫外分光光度计。
②压力蒸汽消毒器(压力为1.1-1.3kg/cm2,相应温度为120-124℃)。
③具塞玻璃磨口比色管。
1.2 试剂
1.2.1 K2S2O8过硫酸钾溶液(5%-m/v):称取25.0gK2S2O8溶于200ml蒸馏水中,加2ml 1mol/L盐酸,稀释至500ml,可使用半年。
1.2.2 NaOH溶液(5%-m/v):称取 12.5g NaOH溶于200ml蒸馏水中,稀释至250.0ml,装于聚乙烯瓶。
1.2.3 盐酸(1mol/L):量取浓盐酸 40.0ml溶于440.0ml蒸馏水(1:11)。
1.2.4 亚砷酸钠溶液(100mg/L):称取100.0ml亚砷酸钠,溶于适量蒸馏水中,稀释至1000ml容量瓶中,使用前临时配制。
1.3 实验操作
1.3.1 工作曲线的绘制
①在八个50ml比色管中,分别取0.0、0.5、1.0、2.0、4.0、5.0、7.0、10.0ug/ml总氮标准使用溶液,用蒸馏水稀释至25ml。
②加入2ml5%K2S2O8溶液,1.0ml5%NaOH溶液,塞紧磨口塞,用纱布及纱绳扎紧管塞,以防蹦出。
③将试管置于压力蒸汽消毒器中,加热40分钟,放气使压力指针回零,乘热加6滴(100mg/l)亚砷酸钠液,并在开水杯中恒温放置5分钟。
④加2.0ml 1mol/L的HCI,冷却后用蒸馏水稀释至50.0ml标线。
⑤在紫外分光光度计上,以蒸馏水作参比,用1cm石英比色皿分别在220nm及275nm波长处测定吸光度,用校准的吸光度绘制工作曲线。见表1。
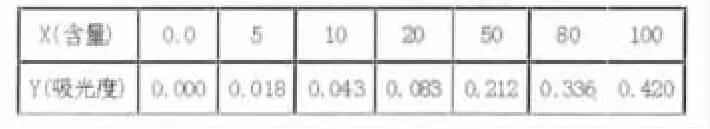
表1 校准曲线的含量及吸光度
1.3.2 水样的测定步骤
取25ml水样(或取适量水样,稀释至25ml)按校准曲线步骤②至⑤操作,然后按校准吸光度(A-A0)计算其总氮浓度。
2 结果与讨论
2.1 K2S2O8-NaOH碱性溶液和K2S2O8-NaOH溶液的稳定性实验
2.1.1 K2S2O8-NaOH碱性溶液不稳定性实验。
常温下,K2S2O8溶液也缓慢地分解
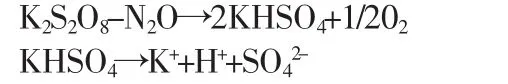
加入NaOH后,OH-与H+结合生成难电离的H2O。因此,加入氢氮化钠后,加速了过硫酸钾的分解。
用新配制的K2S2O8-NaOH碱性溶液,对1.14mg/l的标校做稳定性试验,当天,测试结果准确,误差只有1.8%,第4天浓度降至0.70mg/l,即氧化能力降低了38.5%,10天后,几乎失去了氧化能力。详见表2:
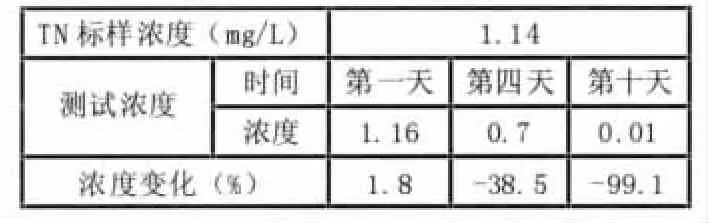
表2 K2S2O8-NaOH碱性溶液稳定性的测定
实际说明K2S2O8-NaOH碱性溶液很不稳定,三天后氧化能力迅速降低。
2.1.2 K2S2O8-HCI溶液的稳定性试验
K2S2O8和NaOH溶液分别配制,并在K2S2O8溶液中加入少量HCI,增加了溶液中H+的浓度,则抑制了K2S2O8的分解。

用K2S2O8-HCI溶液和1.14mg/L国家标样做其稳定性试验,实验表明:从第10天到第60天测定结果无显著性差异,即K2S2O8溶液60天后还有很强的氧化能力,是非常稳定的。
2.2 空白吸光度高的主要原因
将 5%K2S2O81.0ml稀释至 50ml的溶液(K2S2O8溶液20mg/L),以200-275nm扫描测定吸光度发现,S2O82-在220nm的波长上有较高的特征吸收,因此,加热时,K2S2O8没有完全分解是空白吸光度高的重要原因,即是剩余微量的K2S2O8也会引起很高的吸光度。另外K2S2O8不纯也是引起空白吸光度高的一个原因。
2.3 降低空白吸光度的方法
2.3.1 减少K2S2O8和NaOH的用量
标准方法中,K2S2O8原用量是 200mg,NaOH原用量是75mg,改进后K2S2O8、NaOH分别用 100mg和50mg,分别减少 1/2,1/3。
2.3.2 延长消解的时间
标准方法中,原消解时间为30min,为了使K2S2O8充分分解,消解时间40min为宜。当然消解时间更长,会更加降低空白吸光度。
2.3.3 使用二次蒸馏水
配制试剂和实验用水全部用二次蒸馏水效果更佳。
2.3.4 用还原剂亚砷酸钠分解K2S2O8
消解后剩余微量的K2S2O8会影响空白吸光度,加入6滴100mg/l临用前新配制的亚砷酸钠溶液还原微量的K2S2O8。
化学方程式为:
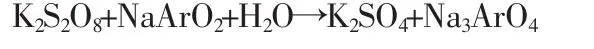
此反应在碱性条件下进行。选用还原剂必须满足三个条件:第一,溶液要无色透明;第二,不含氮元素;第三,在220nm波长上无特征吸收,对 SnCl2、Na2C2O4、抗坏血酸、Na2S2O4和NaAsO2多种浓度不同的还原剂进行了实验,亚砷酸钠有较好的效果。
2.4 测定总氮过程中PH值的变化
在测定TN时,每个环节的PH值是很重要的,特别是加入K2S2O8和NaOH溶液。加热之前,PH值必须在11-13之间,才能有利于K2S2O8的迅速分解。加热40min,PH降至9-11,趁热加入亚砷酸钠,在碱性条件下分解K2S2O8,5min 后,用水冷却。然后加入 2.0mIHCI,且定容至50.0ml时,PH值应小于2。
2.5 改进方法的准确度试验
2.5.1 标准方法与改进方法实验对比
用标准方法和改进方法测定1.14mg/l标样,根据测定浓度的相对误差分析,二者无显著性差异。
2.5.2 测定总氮中NH3-N和NO2-N的转化率
测定总氮时,在加热条件下,K2S2O8的氧化作用,使NH3-N和NO2-N转化为NO3-N,以NH3-N和NO2-N标准溶液做水样测定转化率,其结果转化率分别是98.04%和100.4%,这说明此方法不仅氧化NH3-N和NO2-N为NO3-N有很高的转化率,同时,也有很高的准确度。
2.5.3 标准方法和改进方法加标回收率测定
各取25.0ml地表水样,分别做水样TN加标平行样测定,在地表水中分别加入TN50.0ug按标准方法和改进方法测定总氮的加标回收率,两种方法的加标回收率分别是100.94%和99.52%。
3 结论
3.1 碱性过酸钾溶液,分别配制成K2S2O8-HCI溶液和NaOH溶液,克服了其不稳定的缺点,至少可使用半年。
3.2 S2O82-在220nm波长处有较高的特征吸收,这是测TN空白吸光度高的重要原因,加长消解时间到40min,用二次蒸馏水,降低K2S2O8和NaOH的用量,用低浓度亚砷酸钠溶液还原消解后剩余的微量K2S2O8,可使空白吸光度降低。
3.3 改进方法对NH3-N和NO2-N有很高的转化率,对比试验证明有较高的回收率和准确度,满足监测方法的需要。
[1]董纪珍,宋莹,李建坡,丁致英.测定总氮时影响空白吸光值的因素[J];光谱实验室;2006年04期。
猜你喜欢
辐射防护通讯(2019年3期)2019-04-26
绿色科技(2018年24期)2019-01-19
意林(儿童绘本)(2018年10期)2018-11-08
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
浙江农业科学(2016年11期)2016-05-04
广州大学学报(自然科学版)(2015年4期)2015-12-23
电源技术(2015年2期)2015-08-22
机电信息(2014年35期)2014-02-27
机电信息(2014年26期)2014-02-27
