
讨论乙烷的稳定构象
2010-07-02 00:34:13杨颙梁翰清
大学化学 2010年2期
杨颙 梁翰清
(安徽建筑工业学院材化学院 安徽合肥230601)
讨论乙烷的稳定构象
杨颙 梁翰清
(安徽建筑工业学院材化学院 安徽合肥230601)
利用Gaussian 98对乙烷的两种典型构象——交叉式和重叠式进行结构几何优化、频率分析和能量计算,通过计算结果的比较分析确定其稳定构象;并对导致稳定构象原因进行简单讨论。
乙烷是烷烃中最简单的含碳碳单键的化合物。如果使乙烷中一个甲基固定不动,而使另一个甲基绕碳碳键轴旋转,则两个甲基中的氢原子的相对位置将不断改变,产生许多空间排列方式不同的构象。转动的角度是无穷多的,排列方式也是无穷多的,所以乙烷分子的构象也是无穷多的[1]。交叉式构象和重叠式构象是乙烷无数构象中的两种极端情况。用球棒模型很容易看清楚乙烷分子中各原子在空间的不同排布。如图1所示。

图1 乙烷的两种典型构象
可以利用Gaussian 98通过计算化学的方法对乙烷最具有典型意义的两种极限构象——交叉式和重叠式的稳定性进行比较分析,从而得出何种构象是最稳定构象的结论。
1 研究方法
乙烷交叉式构象和重叠式构象的稳定性可以通过二者能量的大小进行比较判断,能量越低的构象越稳定。基于这个原则,可以进行如下工作:
首先,在HF/6-31G(d)和MP2/6-31G(d)水平下,利用Gaussian 98软件[2]分别对乙烷的交叉式和重叠式构象的分子进行结构几何优化。然后,对所得的计算结果进行分析,比较交叉式和重叠式分子能量的大小,能量低的构象为稳定构象。并进一步通过比较两种构象有无虚频及键长大小等对乙烷稳定构象进行判断。
对于计算方法的选择通常有HF(Hartree-Fock,单组态自洽场从头算)方法和MP2(二级Moller-Plesset微扰理论)方法[3-4]。在自洽场方法中,假定一个电子在由原子核和其他电子形成的平均势场中独立地运动,这只是考虑了粒子之间平均相互作用,但没有考虑电子之间的瞬时相关,即在平均势场中独立运动的两个自旋反平行的电子有可能在某一瞬间在空间的同一点出现。而实际上这是不可能的,因为电子之间存在着库仑排斥。因此,电子实际上并不能“独立”地运动,每个电子在自己的周围建立了一个“库仑穴”[5]。HF方法没有考虑这种自旋反平行的电子相关作用,所以用此法求得的体系能量要比实际高一些,且产生的能量偏差(电子相关能)占体系总能量的比例约为0.3%~1%。而MP2法是一种对电子相关作用进行修正的方法,MP2法类似于多体微扰理论,它将体系分为未微扰体系和微扰体系来考虑。一般地,由于MP2法考虑了电子的相关性,所以MP2法较HF法更为精确。
2 过程、结果和分析
2.1 计算方法
利用GaussianView软件分别构建乙烷分子的两种构象——交叉式和重叠式(图1),并对二者的结构对称性进行设置。先采用基组6-31G(d)和Hartee-Fock方法分别对两构象进行几何优化和频率分析,采用的关键字为:#opt freq hf/6-31g(d)。设置好分子模型后,分别以文件名“c2h6-jiaochashi.gif”和“c2h6-chongdieshi.gif”进行保存。再打开Gaussian 98软件,分别导入文件“c2h6-jiaochashi.gif”和“c2h6-chongdieshi.gif”。所有计算由Gaussian 98软件完成,最后将计算结果保存为文件“c2h6-jiaochashi.out”和“c2h6-chongdieshi.out”。
为了得到更高精度的计算结果,在相同基组上选用MP2方法进行计算。采用的关键字为:#opt freq mp2/6-31g(d)。计算结果保存为文件“c2h6-jiaochashi2.out”和“c2h6-chongdieshi2.out”。在几何优化的基础上,利用关键字“pop=nbo”进一步对乙烷的两种构象进行自然键轨道分析。
2.2 读取结果并分析
用GaussianView软件打开文件“c2h6-jiaochashi.out”和“c2h6-chongdieshi.out”,可以得到几何优化后的分子,如图2所示。

图2 优化后的乙烷的两种构象
用记事本或写 字板打 开“c2h6-jiaochashi.out”、“c2h6-chongdieshi.out”及“c2h6-jiaochashi2.out”、“c2h6-chongdieshi2.out”。可以根据具体需求分别读取 HF方法和MP2方法计算结果,现将两种方法的主要计算结果列于表1。

表1 HF法和M P2法的主要计算结果
通过表1可以看出,HF方法和MP2方法计算结果是类似的。由HF方法的计算结果可知交叉式构象的吉布斯自由能要比重叠式的吉布斯自由能低12.03kJ/mol;而MP2方法的计算结果表明,二者的吉布斯自由能相差约12.51kJ/mol。上述结果可以说明交叉式构象的分子能量要比重叠式构象的分子能量低,这表明乙烷的交叉式构象较重叠式构象稳定。
此外,进一步地从有无虚频以及键长大小方面比较也可以得出相同的结论:一般来说,可以用虚频的数目(0或1)来判断物种是稳定构型还是过渡态,虚频的数目为1的是一阶鞍点,对应的是过渡态的结构。重叠式构象有一个虚频,表明我们找到的是一个连接两个极小值的过渡态结构,而并非真正的稳定结构,可以得出重叠式是不稳定的构象;从表1也可以看出乙烷交叉式构象的C—C键长要比重叠式构象C—C键长短,说明乙烷交叉式构象的C—C键能较大,也就表明乙烷交叉式构象更稳定。
3 讨论
由上述结果可以看出,交叉式为稳定构象,而重叠式则可以看成是一个不稳定的过渡态,从交叉式到重叠式经历了C—C键60°旋转,两种构象的能量差即为旋转活化能。我们用HF法计算得到的旋转活化能约为12.03kJ/mol,而MP2方法的计算结果约为12.51kJ/mol,这与Weinhold计算的乙烷旋转活化能12.54kJ/mol有很好的一致性[6]。另一方面,乙烷交叉式和重叠式构象的能量虽然不同,但能量差并不太大,交叉式转变为重叠式只需吸收约为12.51kJ/mol的能量,即此反应的旋转活化能。反应速率、温度、活化能的关系可用下式进行表示:

其中,k为速率常数,Ea为活化能,R为摩尔气体常量,T为热力学温度,A含有两个因子:频率因子Z(与碰撞频率有关,也叫前因子)和空间因子P(代表可以形成活性复合物的反应物碰撞数在总碰撞中所占的比例,单位与速率常数k单位一致)[7]。
由式(1)可知,温度一定时,Ea值愈小,反应速率愈大。乙烷构象由交叉式转变为重叠式的活化能只有12.5kJ/mol,所以乙烷分子的该转变过程的反应速率很大,常温下乙烷分子在某一构象停留的时间很短(<10-6s)。因此,很难把某一构象“分离”出来。当然,从统计的观点来看,在某一瞬间,乙烷分子中交叉式构象比重叠式构象所占的比例要大得多(在25℃时,每160个交叉式的乙烷才有一个重叠式的乙烷分子)[8]。从式(1)也可以看出,在Ea值一定的情况下,当T值逐渐减小时,反应速率也将逐渐减小,也就是说在低温下,交叉式构象的优势将变得明显,在-170℃时,乙烷基本上都是交叉式构象。
乙烷的交叉式构象较重叠式构象稳定,这一结果已经得到了实验证实,但对产生这种结果的原因却有不同的解释。一般认为重叠式中两个CH3基团上H原子的最近距离为227pm,短于H原子的范德华半径之和(240pm),产生排斥力,从而出现空间阻碍效应;而在交叉型构象中,两个CH3基团中H原子的最近距离为250pm,比两个H原子的范德华半径和要长,不会出现空间阻碍效应的排斥力,能量较低,比较稳定。对乙烷构象的这种解释从1936年提出后,就一直被人们接受,成为化学中的基础内容。但是,周公度先生在《大学化学》上发表文章,认为导致乙烷具有交叉式稳定结构的原因,不是空间阻碍效应的推斥作用所引起,而是超共轭作用所致。超共轭效应是指σ键(常见C—H键)与π键或p轨道,甚至σ键与σ键之间共轭体系表现有某些离域现象。乙烷分子中的超共轭作用就是一种σ-σ超共轭效应,交叉型构象的乙烷分子中,C—Hσ轨道和共平面的另一个CH3基团的C—Hσ*轨道互相叠加,电子产生离域作用,出现超共轭现象的图像示于图3。通过超共轭作用促使能级降低的情况示意见图4[9-10]。
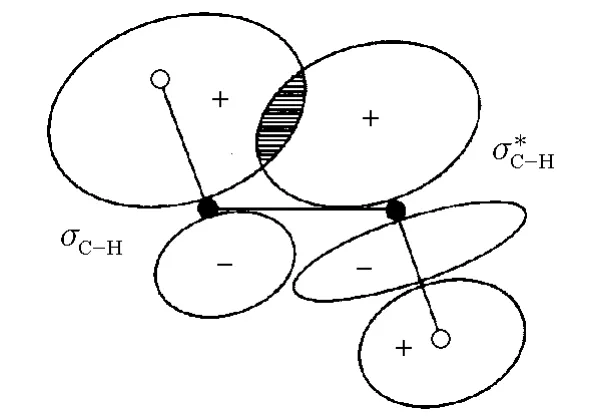
图3 乙烷交叉构象中C—Hσ轨道和C—Hσ*轨道叠加示意图

图4 超共轭作用使体系能级降低的情况
乙烷的交叉式构象较重叠式构象稳定,这种现象也可以用杂化轨道理论进行分析。在MP2/6-31G(d)水平上,利用自然键轨道方法的计算结果,可以分别读出乙烷交叉式构象和重叠式构象C—C键的轨道杂化情况,结果如下。
交叉式构象:s(28.13%);p2.55(71.77%);d0.00(0.10%)
重叠式构象:s(28.01%);p2.57(71.89%);d0.00(0.11%)
由杂化轨道理论可知,乙烷分子中的C—C键是sp3杂化,其中s轨道占25%左右的份额,p轨道占75%左右。s轨道所占的份额越大,键长越小,键能越大,相应的分子就越稳定。而由乙烷交叉式构象和重叠式构象C—C键的分子轨道杂化情况可知:交叉式构象的s轨道所占的份额为28.13%,较重叠式构象的s轨道所占的份额28.01%要大,可以确定乙烷交叉式构象中的C—C键能较大,C—C键较稳定,相应地,乙烷分子中交叉式为稳定构象。
从乙烷的两种构象的对称性来看,交叉式的点群类型为D3d,而重叠式点群类型为D3h,在交叉式向重叠式沿C—C键旋转过程中,点群对称性经历先下降再升高的变化,而能量却一直下降,可以推断对称性不是影响乙烷稳定性的因素。
4 结论
由以上分析可以得出结论:乙烷的交叉式构象较重叠式构象稳定,乙烷的交叉式构象是其优势构象。在室温下,由于乙烷分子中的C—C键能迅速地旋转,构象之间互相转换频繁,因此很难分离出乙烷的某一构象,只是在统计意义上,某一瞬间乙烷分子中交叉式构象比重叠式构象所占的比例要大得多。但在低温时,交叉式构象明显增加,交叉式构象优势明显。
[1] 蔡素德.有机化学.第3版.北京:建筑工业出版社,2006
[2] Frisch M J,Trucks GW,Schlegel H B,et al.Gaussian 98.Pittsburgh:Gaussian,1998
[3] Head-Gordon M,Pople JA,Frisch M J.Chem Phys Lett,1988,153:503
[4] Frisch M J,Head-Gordon M,Pople JA.Chem Phys Lett,1900,166:275
[5] 杨频.分子结构参量及其物性关联规律.北京:科学出版社,2007
[6] Weinhold F.Nature,2001,411:539
[7] 童林荟,申宝剑.超分子化学研究中的物理方法.北京:科学出版社,2004
[8] 伍越寰,李伟昶,沈晓明.有机化学(修订版).第2版.合肥:中国科学技术大学出版社,2002
[9] Pophristic V,Goodman L.Nature,2001,411:565
[10] 周公度.大学化学,2001,16(5):51
猜你喜欢
数学物理学报(2022年1期)2022-03-16 06:15:14
数学物理学报(2021年6期)2021-12-21 06:24:38
煤气与热力(2021年3期)2021-06-09 06:16:22
河北理科教学研究(2020年1期)2020-07-24 08:14:34
应用数学(2020年2期)2020-06-24 06:02:50
石油化工建设(2018年2期)2018-07-11 01:25:04
铜仁学院学报(2018年6期)2018-07-05 09:47:36
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
应用化工(2014年7期)2014-08-09 09:20:23
无机化学学报(2014年8期)2014-02-28 17:32:36
