
磁性纳米材料的化学合成、功能化及其生物医学应用
2010-07-02 00:34侯仰龙
大学化学 2010年2期
侯仰龙
(北京大学工学院先进材料与纳米技术系 北京100871)
今日化学
磁性纳米材料的化学合成、功能化及其生物医学应用
侯仰龙
(北京大学工学院先进材料与纳米技术系 北京100871)
从纳米材料的生长动力学模型出发,讨论磁性纳米材料的控制合成原理。总结磁性纳米材料的化学设计与合成、表面功能化及其在核磁共振成像和多模式影像等方面的应用研究最新进展。
磁性材料在信息存储、传感器和磁流体等传统学科领域有着重要的应用。近年来,随着纳米材料科学与技术的发展,纳米磁性材料的应用开发日益引起人们的关注,特别是在提高信息存储密度、微纳米器件和生物医学领域的应用潜力巨大。本文将从纳米磁学开始,回顾磁性材料的基本概念、化学设计与合成、表面功能化及其在生物医学领域的潜在应用[1]。
1 纳米磁学
在磁场中,铁磁体的磁化强度M或磁感应强度B与磁场强度H的关系可用曲线来表示。当外磁场作周期变化时,铁磁体中的磁感应强度随磁场强度的变化而形成一条闭合线,即磁滞回线,图1(a)为铁磁物质磁滞现象的曲线。一般说来,铁磁体等强磁物质的磁化强度M(或B)不是磁场强度H的单值函数而依赖于其所经历的磁状态。以磁中性状态为起始态,当磁状态沿起始磁化曲线磁化时,此时磁化强度逐渐趋于饱和,曲线几乎与H轴平行,将此时的磁化强度称为Ms。此后若减小磁场强度,则从某一磁场强度开始,M随H的变化偏离原先的起始磁化曲线,M的变化落后于H。当H减小至0时,M并未同步减小到0,而存在剩余磁化强度Mr。为使M减至0,需加一反向磁场,称为矫顽力Hc。反向磁场继续增大时,磁体内的M将沿反方向磁化到趋于饱和(Ms),反向磁场减小至0再施加正向磁场时,按相似的规律得到另一支偏离反向起始磁化曲线的曲线。当外磁场完成如上变化时,铁磁体的磁状态可由图1(a)所示的闭合回线描述。当温度高于居里点时,磁性材料将变成顺磁体,其磁性很容易随周围磁场的改变而改变。如果温度进一步提高,或者磁性颗粒的粒度很小时,即便在常温下,当尺寸达到临界畴时,材料中电子的热运动将逐渐占主导作用,热运动引起的扰动能超过磁能,使得原有的磁有序发生无序化,该现象称为超顺磁现象,如图1(b)所示,此时材料矫顽力和剩磁为0。对于纳米颗粒的超顺磁转变温度,称为Blocking温度。其磁学性质随尺寸的变化,如图2所示,与块体磁性材料的多畴结构相比,纳米颗粒具有单畴结构,当颗粒尺寸小于临界畴尺寸时,纳米颗粒的磁自旋将无序排列。在单畴区域,矫顽力随着颗粒尺寸的增加而增加,在颗粒尺寸大于单畴尺寸时,颗粒呈现多畴结构,只有在一个较小的反向磁场的作用下,其磁化强度才能变为0。磁性材料的临界畴尺寸(Rsd),可用如下公式计算[2]:
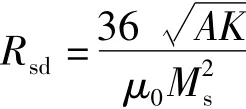
其中,A为交换常数,K为磁晶各向异性常数,Ms为饱和磁化强度。
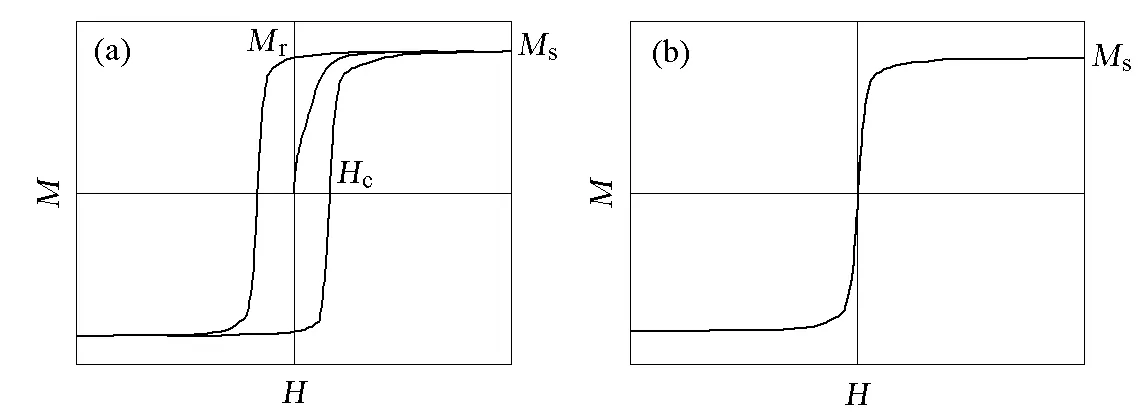
图1 磁滞回线
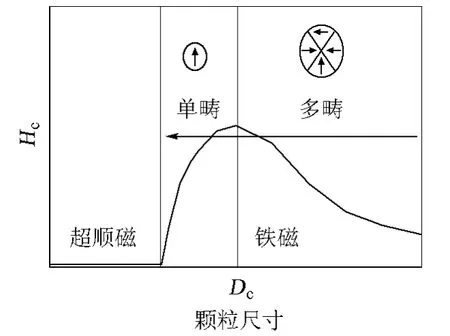
图2 颗粒磁性随尺寸的变化
铁磁性纳米颗粒是理想的磁存储材料,结合垂直磁记录、热辅助图形等技术可大幅提高存储密度。而超顺磁纳米颗粒,因为其相对较弱的磁相互作用,通过相应的表面修饰,在生物体内能够实现良好的分散,因此在药物传输、核磁共振成像和分子探针等领域有重要的应用。
2 纳米颗粒的生长动力学
近年来,已发展出多种化学方法合成高质量的磁性纳米颗粒,包括铁系单质、合金及其化合物等。常见方法有水解沉淀法(包括酸、碱法)、金属有机热分解法、溶胶-凝胶法、微乳液法(W/O)、水热合成法、气溶胶喷射热解法、气相沉积法(CVD)等[3]。本文重点回顾高温有机液相方法合成单分散的磁性纳米颗粒的研究进展。
La Mer理论认为[4],当反应溶液中单体的浓度快速增大并超过超临界浓度时,将快速成核,此后如无新核生成,所形成的核将以同样的速度成长,获得单分散的纳米颗粒,如图3所示[5]。反应过程中,亦有小的颗粒重新溶解到溶液中,以较大的颗粒为核继续长大,最后获得均匀的较大颗粒,即Ostwald熟化。因此,液相合成单分散纳米颗粒的常用技术是分离其成核和生长过程。另外,纳米颗粒因其超大的比表面而容易团聚以减小其表面能。为避免团聚,纳米颗粒表面通常以表面活性剂包覆,表面活性剂间的排斥力通常能够使颗粒得以稳定分散。
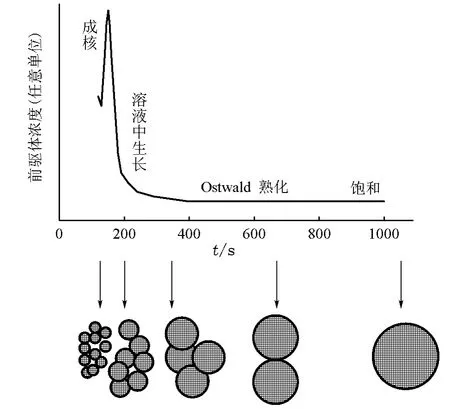
图3 La M er模型示意图及分离胶体颗粒的成核和生长过程[5]
3 磁性纳米颗粒的化学合成
有机金属配合物由于其亚稳态特征,在较温和的条件下,如加热、光照和超声等,可分解成0价的金属,因此常被用作合成磁性金属纳米颗粒的前躯体[6-7]。金属羰基物及其衍生物是一类典型的金属有机配合物,在加热时,羰基很容易与金属核分离,使得0价的金属成核、生长成颗粒。例如,五羰基铁(Fe(CO)5)在油酸保护下,可分解获得单分散的金属铁纳米颗粒;通过八羰基二钴(Co2(CO)8)的分解,可制备单分散的面心立方Co纳米颗粒,而以三烷基膦为稳定剂时,可获得ε-Co纳米颗粒[8]。在1,2-二氯苯中,同时分解Fe(CO)5和Co2(CO)8可以制得FeCo纳米颗粒[9]。值得注意的是,金属纳米颗粒易被空气氧化。为稳定金属纳米颗粒,基于表面活性剂或者壳层结构的表面包覆成为避免深度氧化的重要方法。例如,在制得金属铁的纳米颗粒后,通过弱氧化剂,如N-氧化三甲基胺(Me3NO),控制氧化其表面,形成可控厚度的氧化层,获得核壳型Fe@Fe3O4纳米颗粒[10]。
制备磁性纳米颗粒的另一种常用方法是在表面活性剂的稳定下还原金属盐。与上述热分解过程相比,金属还原方法具有更大的选择性,前躯体可以是金属氯化物、硝酸盐、氧化物、乙酰丙酮盐,还原剂可选用硼氢化钠或超氢锂、多醇、水合肼等。例如,通过三乙基硼氢化锂(超氢)在辛醚中还原CoCl2,利用油酸和三辛基膦的稳定作用,制备了单分散的ε-Co纳米颗粒[8]。通过硼氢化钠同时还原FeSO4和CoCl2,制备FeCo纳米颗粒[11]。在苯甲醚中,油酸和油胺共存时,以LiBEt3H还原FeCl2和Pt(acac)2可以获得4nm FePt颗粒[12]。1,2-烷基二醇常被用作还原剂,用于制备氧化铁的纳米颗粒,例如在苯醚中,利用十六烷基二醇还原Fe(acac)3,可制备单分散的4~18nm Fe3O4纳米颗粒[13],该过程可以扩展到制备铁氧体,包括MFe2O4,(M=Co,Mn)。最近,1,2-十六二醇也被用于在油酸-油胺中还原 Fe(acac)3和Co(acac)2制备FeCo纳米颗粒;合成的20nm FeCo纳米颗粒的饱和磁化率Ms为207cm3·g-1,退火处理,包覆碳层后,其饱和磁化强度达到230cm3·g-1[14]。
烷基胺和酸在升高温度的情况下,也是较强的还原剂。在反应过程中,油酸或者油胺在高温(380℃)时分解,产生一些还原性物质,如C、CO和H2。最近,基于同样的原理,我们仅在油胺和苯甲醚环境中,合成了单分散的Fe3O4纳米颗粒,其尺寸可控制在7~10nm[15],该方法不需要加入价格较高的烷基二醇,而且更为简便实用。
另外,在制备单分散的磁性纳米颗粒时,表面活性剂对纳米晶的成核和生长有较大的影响。通常较短烷基链的稳定作用较弱,纳米晶可以快速生长。相反,较长的烷基链使得纳米晶的生长速度较慢,通常获得较小尺寸的纳米颗粒[5]。
4 形貌各向异性磁性纳米结构的化学合成
除了球形纳米颗粒的合成外,形貌各向异性磁性纳米材料因其形貌依赖磁学行为等特性,引起了人们的关注。利用烷基胺和烷基酸的还原化学,纳米立方和中空的纳米框等各向异性纳米结构得以合成[16],中空结构的产生主要源于熔盐的腐蚀。在Co表面氧化和快速扩散时,亦可获得中空CoO纳米颗粒。在油酸-油胺体系中,于300℃还原Fe(acac)3,可获得FeO纳米颗粒和纳米立方体(图4(a))[17]。其形貌的控制主要依赖于表面化学,在油酸和油胺摩尔比小于1时,稍过量的油胺起主导作用,因其对氧化铁表面的稳定作用相对较弱,使得纳米晶在各个晶向的生长速度相近,产物即成球形颗粒。而在油酸过量时(两者摩尔比大于1),羧酸根对氧化铁表面具有较强的稳定作用,只有特殊晶面得以优先生长,这样就获得了形貌各向异性的纳米立方体。通过改变反应条件,在油胺中也得到了一维FePt纳米线和纳米棒,其长度可以控制在20~200nm(图4(b))[18-19];初步实验研究表明,其生长机理可能是油胺在纳米晶的生长过程中形成准一维的空腔结构,类似于软模板,前躯体在该空腔结构中分解生长,获得一维结构。当引入第二种溶剂时,由于油胺浓度降低,相当于剪裁了空腔的纵向长度,获得了单分散的纳米棒。最近,利用类似的体系,合成了单分散的22nm Fe3O4纳米八面体(图4(c),(d))[20]。

图4 形貌各向异性的磁性纳米结构
5 异质磁性纳米结构
多功能异质纳米颗粒,因其多元组分提供了多功能的界面与功能,在纳米催化和生物医学领域具有巨大的应用潜力,引起了人们的关注和兴趣。目前,通常有两种策略用于构建异质磁性纳米颗粒。一是分子功能化,如链接抗体、蛋白和染料等;另一种方法是整合磁性纳米颗粒与其他功能化的颗粒于一体,如链接量子点、金属颗粒等[21]。磁性纳米颗粒与量子点结合,使得多功能纳米颗粒具有磁学和光学的性质。而与金属纳米颗粒的复合可以获得等离子发光的特性。此外,其复合的多元结构还可能为药物治疗和传输提供平台,因为多功能纳米颗粒不仅具有增强的功能,还具有功能的多样性,所以在生物医学领域具有独特的应用优势。本节重点回顾基于第二策略的多功能纳米颗粒合成的最新进展。
异质结构和球壳结构一样,通过将几种不同功能的组分结合在一起,使其作为一个多功能体。不同点在于异质结构的不同组分都暴露在外侧,从而显示出一定的各向异性。目前,在生物探针领域,异质结构以二聚体为主。量子点因其独特的光学稳定性和优于荧光染料的抗光淬灭能力,在纳米生物医学的基础研究中得到广泛的应用[22]。此外,量子点在体内的影像研究中具有多选择性。基于FePt纳米颗粒和硫族半导体纳米组分的复合纳米颗粒的系统研究表明,反应条件控制着不同的杂化结构的形成。在一步反应中,依次向FePt纳米颗粒的溶液中加入CdX(X=S or Se),低温时形成FePt@CdX核壳纳米颗粒(如图5(a),(c))[23-24]。然而,用高沸点溶剂时,得到FePt-CdX二聚体纳米颗粒(图5(b),(d))。在高温时,二聚体的形成可能是因为FePt和CdX间不同的相转变温度所致。CdX在高温时可能熔融,引起与FePt核的剥离。这些核壳和二聚体结构的颗粒的合成具有很好的重复性,虽然其光学性质由于猝灭等原因需要进一步改进,但是其合成过程简单,便于操作。

图5 核壳和异质二聚体纳米结构[23-24]
6 磁性纳米聚集体
磁性纳米聚集体在生物医学的蛋白分离和诊断中具有广泛的应用,通常由磁性纳米颗粒和高分子骨架材料制备而成。其中高分子材料包括硅烷、聚丙烯酸、淀粉、葡聚糖、明胶、乙基纤维素等。制备磁性纳米聚集体的方法可分为直接法和间接法:直接法是在成球前即加入磁性纳米材料,成球时聚合物将其包裹其中或粘附其外;间接法是先制备非磁性小球,然后通过处理使磁性材料进入其中,磁性纳米粒以分散的形式存在于微球的骨架材料中。目前,国内外对磁性纳米聚集体的研究取得了一些重要的研究成果。在最近的工作中,我们利用乙二醇作为溶剂和还原剂,以乙酰丙酮铁为前躯体,通过环糊精特有的空腔结构的螯合作用,自组装合成了尺寸可控的磁性纳米微球[25],其尺寸可调控在20nm和几个微米。以简单氯化物和醋酸钠为前躯体,也可合成200~800nm左右的铁氧体微球[26]。
7 生物医学应用
纳米技术与分子生物学相结合发展出一个新的研究领域——纳米生物技术。磁性纳米颗粒是一类具有可控尺寸、能够外部操控并可用于核磁共振成像(MRI)造影的材料。这使得该类纳米颗粒能够被广泛应用于生物学和医学领域,包括蛋白质提纯、药物传输和医学影像等方面[27]。当纳米颗粒与靶向试剂耦合,通过特定的生物作用与生物分子反应,功能化的纳米颗粒即可与靶向生物组织耦合,实现疾病诊断或者生物分离。在核磁共振成像的正常磁场强度下(通常高于1T),这些靶向区域的超顺磁纳米颗粒可以达到磁饱和,建立有序的区域扰动偶极场,缩短MRI中质子弛豫时间,使得靶向区域相对于生物环境有更暗的对比度。此外,在可控场幅和频率的交变磁场中,链接于生物体的超顺磁纳米颗粒的磁矩可以翻转,磁矩的再取向使得纳米颗粒与其周边的生理环境之间或是磁易轴与原子内部晶格间产生了“摩擦”,源于这种“摩擦”的能量转化为热能,使得这些超顺磁纳米颗粒可用作热源加热靶向区域,即实现磁流体热疗,被广泛用于研究癌症治疗。以下将介绍磁性纳米颗粒的表面功能化方法和几类材料的生物医学应用研究进展。
7.1 磁性纳米材料的表面功能化
由于其尺寸和形貌以及化学组成的均一性,有机相制备的高磁矩纳米颗粒是一类理想的生物医用材料。然而,合成的超顺磁纳米颗粒表面通常被油酸和油胺等表面活性剂包覆,为疏水表面。为使这些纳米颗粒水溶而用于生物医学,其表面经常需要进行功能化,主要策略有两种,增加表面层或者替换表面活性剂(配体),如图6所示。配体替换时利用新的双功能配体直接替换原来的配体,该双功能分子一端具有与颗粒表面有强键合作用的基团,另一端是可以使纳米颗粒水溶或者可进一步功能化的极性基团(图6路径(a))。配体叠加是通过包含疏水和亲水基团的双亲分子实现的,疏水基团与原来的碳氢链形成双层结构,亲水基团暴露在颗粒的表面使其具有水溶性(图6路径(b))。
Dai等发展了聚乙二醇(PEG)修饰的磷脂微囊,用以修饰FeCo纳米颗粒或中空MnO纳米颗粒[28],磷脂的碳氢链通过疏水相互作用,链接到颗粒表面的碳层形成稳定的双层,同时PEG暴露在水中实现很好的水溶性。相似的双亲高分子,如聚苯乙烯-聚丙烯酸嵌段共聚物(PSPAA),十四烷基磷酸酯和聚乙二醇-2-十四烷基乙醚等也被用于转化非水溶性纳米颗粒为水溶性[29]。与双亲微囊包覆相比,从原理上讲,双功能配体替代,因为其双齿或多齿结构与颗粒表面的相互作用,使其在生理环境中具有更好的稳定性。Xu等报道了双齿分子,如多巴胺,因为其五元环的轨道重叠和减小的空间效应,能够与氧化铁表面进行有效键合,如图7所示[30]。多巴胺与PEG复合使用,不仅增强了纳米颗粒在水中的稳定性,而且提供了一个简单的方法用于稳定生物分子和磁性纳米颗粒。胺基端修饰的PEG配体用以稳定Fe3O4纳米颗粒,很容易与不同的分子(如色酮)链接,可用于药物传输[31]。
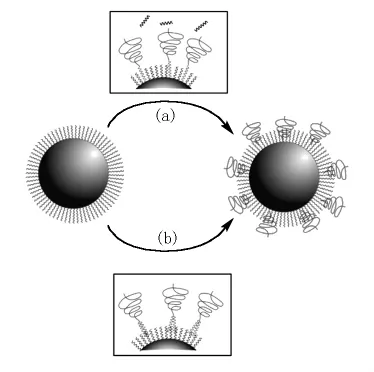
图6 纳米颗粒的表面修饰策略
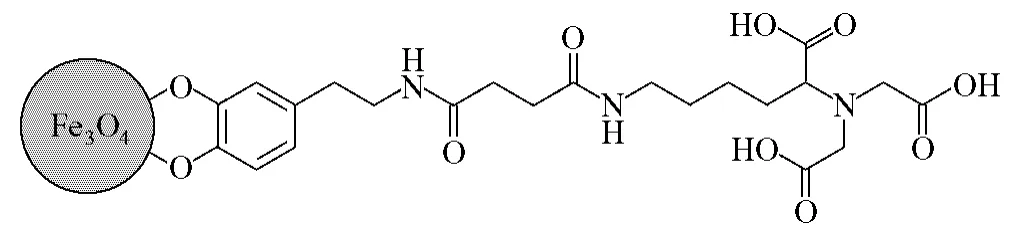
图7 多巴胺端基修饰的Fe3O4纳米颗粒[30]
配体替换策略也适用于有机硅烷替代颗粒表面的疏水配体。基于脱水和酯化作用,有机硅烷中的烷氧基与金属氧化物表面形成共价键相互作用。研究发现,胺基、羧酸基和聚乙二醇为端基的硅烷不仅使磁性纳米颗粒高度稳定、水溶,而且具有多功能性,可用于链接生物分子。基于金属-硫键或者螯合作用,胱胺和2,3-二巯基琥珀酸(DMSA)可替代疏水配体[32],与PEG或者其他高分子修饰的纳米颗粒不同,该类修饰的纳米颗粒的动力学直径相对较小。
此外,一步包覆制备水溶性磁性纳米颗粒近年来获得了较好进展。通常磁性纳米颗粒在不同的羧酸基团,如柠檬酸、磷酸或者其他多齿配体中,获得动力学稳定的水溶性磁性纳米颗粒。Xie等发展了一种新方法用于制备超小的磁性纳米颗粒,其中小配体4-甲基苯膦二酚(4-MC)用作表面活性剂来稳定颗粒的表面,与多巴胺相似,其与氧化铁表面也具有很强的螯合作用[33],进而利用Mannich反应耦合,与环状多肽(c(RGDyK))链接,可用于靶向诊断αvβ3-富集的肿瘤细胞。
7.2 超顺磁纳米颗粒的MRI造影剂
基于质子沿着外加磁场排列和旋动原理设计而成的核磁共振成像(MRI)是临床广泛使用的无损伤诊断模式。当施加一个横向脉冲磁场时,沿着磁场排列的质子产生旋转扰动而偏离磁场方向。当撤除脉冲磁场后,质子偶极矩逐渐回复到原来的位置,即弛豫过程。两个非依赖的弛豫过程(纵向弛豫(T1)和横向弛豫(T2)),分别用于产生一个亮场和暗场MR图像。弛豫的区域变化(即对比度)取决于质子密度和组织器官的化学和物理特征。通过组织中超顺磁纳米颗粒的富集,使其成为典型的T2造影剂用以获得暗场图像,其对比增强正比于磁场强度[3]。
超顺磁纳米颗粒在特定磁场中的磁场强度与颗粒的大小、磁晶各向异性常数有关[3,5],因此核磁共振成像效果亦依赖于超顺磁纳米颗粒的尺寸和结构。最近的铁氧体MRI应用研究发现[34],12nm MnFe2O4具有最好的磁化强度110cm3·g-1,而12nm的 Fe3O4、CoFe2O4和NiFe2O4的磁化强度分别为101、99和85cm3·g-1。每个样品在1.5T磁场下的自旋-自旋弛豫时间(T2)-权重的MRI对比度与其磁性相对应,MnFe2O4纳米颗粒具有最强的对比度,其弛豫率达到 358L·mmol-1·s-1,远高于 Fe3O4,CoFe2O4和 NiFe2O4纳米颗粒的 218,172和152L·mmol-1·s-1。图8表示的是Fe3O4和MnFe2O4的尺寸和结构与其MRI弛豫率的关系,由图8可见,大颗粒具有高的对比效应,但是同一尺寸的纳米颗粒中,MnFe2O4纳米颗粒因其最小的磁晶各向异性常数而磁轴容易反转,具有最高的造影增强效应。在最新的研究中,评价了这些铁氧体纳米颗粒的癌症诊断敏感性。在实验中,MnFe2O4纳米颗粒与具有肿瘤靶向功能的赫塞汀(Herceptin)耦合,通过HER2/neu的耦合作用,可实现在乳腺癌和卵巢癌的表面超表达。结果表明,经MnFe2O4-Herceptin纳米颗粒处理的癌症细胞,在彩色MRI图像中颜色从红色变为蓝色。与之相比,在同一实验中,Fe3O4-Herceptin纳米颗粒处理的癌症细胞没有明显的变化。由此可见,MnFe2O4-Herceptin耦合体具有较高MR灵敏性,可用于肿瘤诊断。此外,利用大磁矩的超顺磁纳米颗粒可设计更高灵敏度的MRI探针,FeCo基纳米颗粒的弛豫率r2达到644L·mmol-1·s-1[35]。
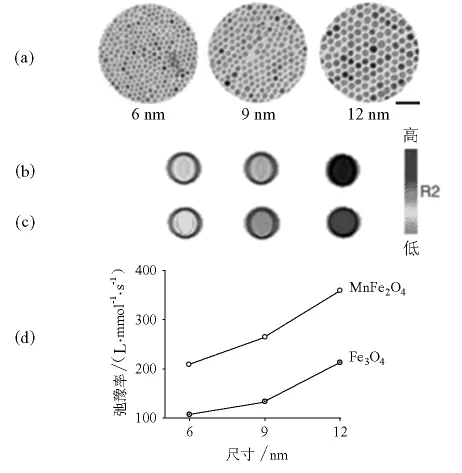
图8 M nFe2O4和Fe3O4纳米颗粒的尺寸与MRI效果[34]
值得一提的是,超小Fe3O4纳米颗粒(动力学直径<10nm)在癌症的早期诊断有着特别的应用价值。最近研究表明,4.5nm Fe3O4纳米颗粒作为核,并与环状多肽(c(RGDyK))耦联[36],通过载有U87MG肿瘤的小鼠T2-自旋回声MRI,评价了体内c(RGDyK)-Fe3O4纳米颗粒与整联蛋白αvβ3的耦合能力。c(RGDyK)-Fe3O4纳米颗粒的r2弛豫率为165L·mmol-1·s-1,这大于同尺寸商业化的菲立磁(Feridex)纳米颗粒的数值(104L·mmol-1s-1)。在注射c(RGDyK)-Fe3O4纳米颗粒后,肿瘤的MR信号强度明显降低,该变化在c(RGDyK)(10mg· kg-1)阻断作用下得到明显抑制,这一结果证实了与多肽链接的纳米颗粒的靶向性。
7.3 多功能分子探针
核壳结构复合微粒,其内部和外部分别富集不同成分,显示出特殊的双层或者多层结构,通过核与壳的功能复合与互补,调制出有别于核或壳本身性能的新型功能材料,为新材料的设计提供了非常便捷的途径。因此,通过核壳结构很容易得到所需的多功能分子探针。在各种核壳结构中,基于氧化硅的材料比较多见,这也是由于氧化硅的外壳制备工艺简单,易通过修饰实现功能化。二氧化硅包覆的四氧化三铁核壳结构,通过APS修饰后,再引入FITC作为荧光基团,可用作神经胶质瘤诊断的荧光/MRI多功能探针[37]。此外,氧化钆纳米颗粒是一种较好的核磁共振T1成像造影剂,也可与荧光分子链接制备双功能探针。例如,用TEOS和APS耦联的荧光染料将所得Gd2O3纳米颗粒包覆了一层荧光性氧化硅壳,并通过体内体外实验表征所得探针的有效性[38-39]。
量子点作为一种很好的荧光材料,在生物探针领域有较好的应用,但是其毒性问题一直影响其应用。通过二氧化硅的包覆,使之与机体隔绝,是一种较好的解决方法。用二氧化硅包覆量子点类发光材料,并在二氧化硅壳的表面修饰DOTA,使之与钆离子结合,可制备荧光/核磁多功能探针,用于细胞内输运、光敏感性、微循环效应研究[40]。CdSe纳米颗粒是一种性质优越的荧光材料,二价锰离子是一种核磁共振T1造影剂,通过在CdSe纳米颗粒表面包覆一层锰掺杂的硫化锌,可制备一种基于量子点核壳结构的荧光/MRI双功能探针[41]。
金的化学性质稳定,容易修饰,是很好的光学成像探针,以金为外壳的核壳结构受到了广泛的关注。Hyeon等人设计了一种新型的核壳结构,首先制备了表面为胺基氧化硅的纳米颗粒[41],通过有机反应将Fe3O4纳米颗粒与Au纳米颗粒链接到所制得的氧化硅表面,在一定条件下,以金纳米颗粒为晶核使其继续生长,最终得到SiO2-Fe3O4@Au核壳结构,并验证了作为双功能探针的有效性。
除了核壳结构纳米颗粒外,还开发了兼具多种表面特性的二聚体纳米颗粒并应用于分子探针研究。最近,Cheon等人在FePt的纳米颗粒基础上,通过还原金的配合物[42],成功制备了FePt-Au二聚体。使用端基为二巯基的聚乙二醇将所得纳米颗粒转移到水中,并进一步分别修饰了中性链亲和素和HmenB1抗体,并通过后者验证了其作为MRI探针的有效性。Hyeon等人将这个体系拓展到多种材料,分别研究了金、银、铂、镍等金属纳米颗粒、Fe3O4纳米颗粒以及MnO纳米颗粒的复合结构[43],并选择Au-Fe3O4纳米颗粒体系,研究了其光谱性质与MRI成像效果。Sun研究组采用不同的表面化学方法来修饰异质结构的不同部分[44],以多巴胺为耦联剂,将Fe3O4纳米颗粒一侧修饰了带有表皮生长因子抗体的聚乙二醇长链,在另一侧通过巯基为耦联剂修饰了与前边不同长度的聚乙二醇长链;以A431细胞作为研究对象,探讨了该双功能探针的有效性。Mou等人研究了另外一类异质结构,首先制备了Fe3O4@SiO2核壳结构[45],将所得纳米颗粒与FITC作为前驱体来制备多孔二氧化硅,得到Fe3O4@SiO2-多孔SiO2二聚体,并通过大鼠骨髓基质细胞验证了所得纳米二聚体的双功能探针性能。
7.4 纳米聚集体分子探针
纳米聚集体是指通过简单的化学键或分子间作用力,将两种或两种以上的功能组分联结起来的结构。Cheon等人制备了表面为氨基的含有染料的二氧化硅纳米颗粒[46],以及表面为巯基的Fe3O4纳米颗粒,通过耦联剂(SMCC)将两者链接,得到了以SiO2为核心的卫星状结构。通过对比研究发现,这种卫星状结构由于含有较大密度的纳米颗粒,其MRI信号强度有较大提高。将所得纳米颗粒耦联HmenB1抗体,以CHP-134和HEK293T细胞作为研究模型,讨论了其作为双功能探针的作用。Haam等人将Fe3O4纳米颗粒与阿霉素包埋到聚乳酸/乙醇酸共聚物纳米颗粒中[47],以阿霉素作为荧光基团,Fe3O4纳米颗粒作为MRI探针,在纳米颗粒表面进一步修饰Herceptin,并研究了该探针的靶向性。在进一步的工作中,通过纳米乳液法,将荧光分子芘修饰的PCL-PMMA嵌段共聚物与MnFe2O4纳米颗粒复合制备了双功能纳米探针,并进一步用爱必妥(Cetuximab)单抗修饰[48]。通过A431和MCF7细胞,系统地研究了该探针与EGFR的靶向作用,验证了所得荧光/MRI双功能探针的有效性。Hyeon等人利用阴离子表面活性剂在水中形成胶束的现象,包裹聚(D,L-胶乳-联-羟基乙酸)(PLGA)、Fe3O4纳米颗粒、量子点与阿霉素为一体[49],通过蒸发将胶束中的有机溶液去除,得到纳米聚集体,然后又通过叶酸修饰的阳离子表面活性剂将叶酸链接到所得纳米颗粒上。通过叶酸作为靶向试剂,测试了所得纳米颗粒作为双功能探针以及药物输送载体的效果。
8 结论
以上简要介绍了纳米磁学的基本概念和高质量纳米材料的设计与控制原理,介绍了纳米材料化学合成的最新进展;讨论了纳米材料的形貌和结构控制的基本思路以及超顺磁纳米材料的稳定与修饰方法;概述了生物医学应用中MRI成像造影剂增强剂基本原理以及基于不同异质结构的分子探针的设计和应用进展。这些进展证实,基于表面化学控制晶体生长的化学方法而合成的磁性纳米颗粒在高灵敏度医学诊断、高效治疗等领域具有重要的研究价值和应用潜力。
[1] Cullity B D.Introduction to Magnetic Materials.Reading MA:Addison-Wesley Publishing Company,1972
[2] Skomski R,Coey JM D.PermanentMagnetism.Bristol and Philadelphia:Institute of Physics Publishing,1999
[3] Frey N A,Peng S,Cheng K,et al.Chem Soc Rev,2009,38:2532
[4] La Mer V K,Dinegar R H.JAm Chem Soc,1950,72:4847
[5] Murray C B,Kagan C R,Bawendi M G.Annu Rev Mater Sci,2000,30:545
[6] Hyeon T.Chem Commun,2003,927
[7] Cushing B L,Kolesnichenko V L,O'Connor C J.Chem Rev,2004,104:3893
[8] Murray C B,Sun S,Doyle H,et al.MRSBull,2001,26:985
[9] Hütten A,Sudfeld D,Ennen I,et al.JMagn Magn Mater,2005,293:93
[10] Peng S,Wang C,Xie J,et al.JAm Chem Soc,2006,128:10676
[11] Huang J,He L,Leng Y,et al.Nanotechnology,2007,18:415603
[12] Sun S,Anders S,Thomson T,et al.JPhys Chem B,2003,107:5419
[13] Sun S,Zeng H,Robinson D B,et al.JAm Chem Soc,2004,126:273
[14] Chaubey G S,Barcena C,Poudyal N,et al.JAm Chem Soc,2007,129:7214
[15] Xu Z,Shen C,Hou Y,Chem Mater,2009,21:1778
[16] Kim D,Park J,An K,et al.JAm Chem Soc,2007,129:5812
[17] Hou Y,Xu Z,Sun S.Angew Chem Int Ed,2007,46:6329
[18] Sun S,Murray C B,Weller D,et al.Science,2000,287:1989
[19] Wang C,Hou Y,Kim J,et al.Angew Chem Int Ed,2007,46:6333
[20] Zhang L,Wu J,Liao H,et al.Chem Commun,2009,4378
[21] Gao J,Gu H,Xu B.Acc Chem Res,2009,42:1097
[22] Michalet X,Pinaud F F,Bentolila L A,et al.Science,2005,307:538
[23] Gu HW,Zheng R K,Zhang X X,et al.JAm Chem Soc,2004,126:5664
[24] Gu HW,Zheng R K,Liu H,et al.Small,2005,1:402
[25] Hou Y,Gao S,Ohta T,et al.Eur J Inorg Chem,2004,1169
[26] Deng H,Li X,Peng Q,et al.Angew Chem Int Ed,2005,44:2782
[27] Corot C,Robert P,Idee JM,et al.Adv Drug Delivery Rev,2006,58:1471
[28] SeoW S,Lee JH,Sun X,et al.Nat Mater,2006,6:971
[29] Kim SW,Kim S,Tracy JB,et al.JAm Chem Soc,2005,127:4556
[30] Xu C,Xu K,Gu H,et al.JAm Chem Soc,2004,126:9938
[31] Wang B,Xu C,Xie J,et al.JAm Chem Soc,2008,130:14436
[32] Jun YW,Huh Y M,Choi JS,et al.JAm Chem Soc,2005,127:5732
[33] Xie J,Chen K,Lee H Y,et al.JAm Chem Soc,2008,130:7542
[34] Lee JH,Huh Y M,Jun YW,et al.NatMed,2007,13:95
[35] SeoW S,Lee JH,Sun X,et al.Nat Mater,2006,6:971
[36] Meng X,Wan J,Jing M,et al.Acta Pharmacol Sin,2007,28:2019
[37] Bridot JL,Faure A C,Laurent S,et al.JAm Chem Soc,2007,129:5076
[38] Gerion D,Herberg J,Bok R,et al.JPhys Chem C,2007,111:12542
[39] Bakalova R,Zhelev Z,Aoki I,et al.Bioconjugate Chem,2008,19:1135
[40] Wang S,Jarrett B R,Kauzlarich SM,et al.JAm Chem Soc,2007,129:3848
[41] Kim J,Park S,Lee JE,et al.Angew Chem Int Ed,2006,45:7754
[42] Choi J,Jun Y,Yeon S,et al.JAm Chem Soc,2006,128:15982
[43] Choi SH,Na H B,Park Y I,et al.JAm Chem Soc,2008,130:15573
[44] Xu C,Xie J,Ho D,et al.Angew Chem Int Ed,2008,47:179
[45] Wu SH,Lin Y S,Hung Y,et al.ChemBioChem,2008,9:53
[46] Lee JH,Jun Y W,Yeon S I,et al.Angew Chem Int Ed,2006,45:8160
[47] Yang J,Lee C H,Park J,et al.JMater Chem,2007,17:2695
[48] Yang J,Lim E K,Lee H J,et al.Biomater,2008,29:2548
[49] Kim J,Lee JE,Lee SH,et al.Adv Mater,2008,20:478
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
军事文摘·科学少年(2017年4期)2017-06-20
材料科学与工程学报(2016年2期)2017-01-15
现代检验医学杂志(2016年3期)2016-11-15
三峡大学学报(自然科学版)(2016年6期)2016-04-16
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
物理实验(2015年9期)2015-02-28
应用化工(2014年8期)2014-08-08
无机化学学报(2014年4期)2014-02-28
储能科学与技术(2014年6期)2014-02-27
