
侧脑室注射STZ对大鼠学习记忆障碍及海马tau蛋白AD特异性位点磷酸化增加的影响
2010-06-29 07:53第五永长辛馥伶苗迎春
中西医结合心脑血管病杂志 2010年8期
第五永长,辛馥伶,李 敏,赵 娜,苗迎春,时 晶
阿尔茨海默病(Alzheimer's disease,AD)是一种以进行性记忆认知损害和智能障碍为主的中枢神经系统退行性疾病。目前公认的AD核心病理特征为大脑局部,尤其是海马和皮层神经元退行性病变,细胞内神经原纤维缠结(NFT)和细胞外老年斑(SP)沉积。然而,AD的病因和确切发病机制目前仍不清楚。葡萄糖代谢紊乱和能量生成减少与淀粉样蛋白沉积以及tau蛋白过度磷酸化密切相关,可能促进了AD的发生。链脲佐菌表(STZ)侧脑室注射已被证实可以引起大鼠脑持久的葡萄糖代谢紊乱、能量生成障碍伴海马乙酰胆碱转移酶活性降低和氧化应激反应以及学习记忆障碍[1],然而其与AD病理改变的联系未得到进一步实验证实。本实验进一步观察了STZ侧脑室注射对大鼠学习记忆及海马组织tau蛋白AD特异性磷酸化位点Ser422的磷酸化程度的影响。
1 材料与方法
1.1 动物及分组 SPF级健康雄性Sprague-Dawley(SD)大鼠36只,体重(250±20)g。随机分成假手术组、STZ侧脑室注射模型组,每组18只。实验过程中动物自由摄食和饮水(术前禁食除外),室温22℃ ~ 26℃,湿度为28%~30%。
1.2 药品与试剂 链脲佐菌素:Sigma公司;抗体:pS214(western blot检测及免疫组化检测的一抗分别购自Biosource和Abcam公司)。
1.3 ICV-STZ拟 AD模型制备 参考 Sharma等[2]方法,所有大鼠经10%水合氯醛(4 mL/kg)麻醉后,固定于江湾Ⅰ型C大鼠立体定位仪上,常规消毒皮肤,正中矢状切口,分离骨膜,锥颅器钻开颅骨,暴露硬脑膜。除假手术组外,其余动物均用微量注射器每侧侧脑室各注射 STZ约 18 μ L(3 mg/kg,注射前将STZ溶于人工脑脊液,浓度为25 mg/mL)。坐标参考《大鼠脑立体定向图谱》[3]:前囟后1.5 mm,矢状缝左右旁开1.5 mm,脑表面下3.5 mm。第3天重复注射,剂量同前。假手术组以等量人工脑脊液替代STZ。术后常规饲养21 d后行为学测试并处理第一批,31 d后行为学测试并处理第二批动物。
1.4 学习记忆能力测试 采用Morris水迷宫试验法。
1.5 脑组织样品处理 于行为学测试完毕后,分别于、两个时间点选取动物,灌注固定,按4 mL/kg的剂量腹腔注射10%水合氯醛麻醉大鼠。迅速插导管于左心室至升主动脉并固定,同时剪开右心耳。快速灌注生理盐水100 mL至肝脏完全变白,右心室流出澄清液体后,更换4%多聚甲醛先快后慢继续灌注30 min,然后断头,完整取出鼠脑,置固定液中,24 h后石蜡包埋切片。每只大鼠海马CA1区出现后连续冠状切片,放入0.01 mol/L PBST的溶液中进行免疫组化染色。
Western-blot蛋白印迹检测取用新鲜脑组织置液氮中速冻后保存于-70℃冰箱。
2 结 果
2.1 行为学测试结果(见表1、表2)
表1 造模21 d后两组定位航行试验结果(±s)

表1 造模21 d后两组定位航行试验结果(±s)
组别 n 平均逃避潜伏期(s)第21天 第22天 第23天游泳距离(cm)第21天 第22天 第23天假手术组 6 57.82±9.86 40.36±13.06 19.49±11.98 703.15±191.96 521.56±218.58 86.21±139.44模型组 9 107.23±14.961) 96.14±25.261) 79.26±18.161) 1 261.83±314.351)1 039.38±229.341)756.38±196.551)与假手术组比较,1)P<0.01
表2 造模31 d后两组定位航行试验结果(±s)

表2 造模31 d后两组定位航行试验结果(±s)
组别 n 平均逃避潜伏期(s)第31天 第32天 第33天游泳距离(cm)第31天 第32天 第33天假手术组 6 86.93±17.98 54.90±19.39 19.77±12.87 814.72±135.22 589.20±214.47 234.55±134.94模型组 8 113.95±17.221) 103.05±24.901) 67.23±14.551) 853.36±168.201) 830.42±256.291) 619.84±148.411)与假手术组比较,1)P<0.01
2.2 免疫组化检测结果(见表3)
表3 造模第21天、第31天后大鼠海马CA1区tau-ps422表达(±s)

表3 造模第21天、第31天后大鼠海马CA1区tau-ps422表达(±s)
组别 第21天后阳性目标平均数 阳性目标总面积 阳性目标平均灰度值第31天后阳性目标平均数 阳性目标总面积 阳性目标平均灰度值假手术组 2.61±0.12 16.19±2.03 8.40±3.11 4.01±1.05 19.87±4.12 13.85±4.16模型组 11.36±2.911) 30.61±3.252) 20.72±3.862) 20.58±4.121) 51.36±5.112) 38.62±5.282)与假手术组比较,1)P<0.01,2)P<0.05
2.3 Western blot检测结果(见表4)
表4 造模第31天后大鼠海马区 tau-ps422表达(±s)
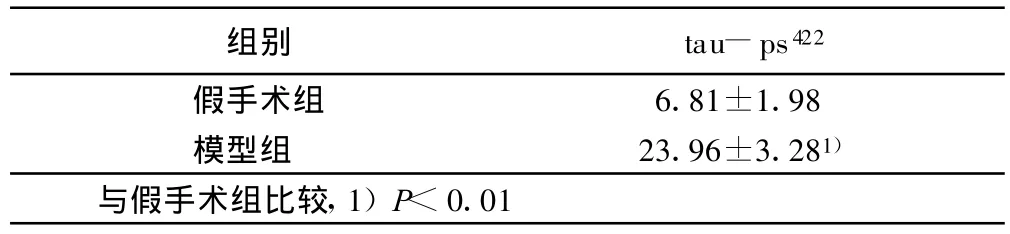
表4 造模第31天后大鼠海马区 tau-ps422表达(±s)
组别 tau-ps422假手术组 6.81±1.98模型组 23.96±3.281)与假手术组比较,1)P<0.01
3 讨 论
STZ是一种甲基亚硝基脲产物,具有抗菌、抗肿瘤的性能和致糖尿病的副反应。1996年Hoyer带领的研究小组首次用侧脑室注射STZ的方法来干扰胰岛素/受体(Ins/IR)信号系统,使脑内出现渐进性葡萄糖和糖原合成减少[4]。而有实验证明Ins/IR信号转导系统障碍是引起AD脑内葡萄糖/能量代谢异常的主要原因[5],在此基础上形成糖基化终末产物(AGEs),并最终导致Aβ及NFT的形成[6]。2005年美国布朗医学院的dela Monte教授和她的研究小组验证了AD是“3型糖尿病”的假说。通过对尸检脑组织进行检测,发现AD患者脑内胰岛素、IGF及其受体的蛋白含量和基因表达均较对照组明显降低;2006年该小组通过对小鼠侧脑室注射STZ的方法阻断胰岛素受体自身磷酸化和内在的酪氨酸激酶活性[7],损伤胰岛素和IGF信号转导通路,实验发现,微量的STZ未引起外周血糖升高,胰腺结构和胰岛素的免疫活性也没受到影响;却出现脑体积缩小,细胞丢失、神经胶质增生、Aβ沉积增多、tau蛋白过度磷酸化、泛素化等惊人变化,学术界一致认为该模型成功地模拟了SAD的病理特征。目前认为,STZ破坏Ins/IR信号转导通路,降低了大鼠脑内葡萄糖利用(GU),引起脑能量代谢障碍,ATP、GTP生成减少,造成认知功能相关皮层的神经细胞功能受损,影响突触传递和突触可塑性,导致学习记忆功能障碍,这也是引起基底前脑合成乙酰胆碱减少、氧化应激、轴突和突触损伤的基础。近年进一步研究认为,大脑葡萄糖代谢低下可通过改变tau的O-GlcNAc糖基化修饰调节tau的磷酸化,在AD发病机制中起着重要作用。与脑葡萄糖能量代谢障碍同时存在的葡萄糖转运体GLUT1与GLUT3减少和tau蛋白的O-GlcNAC糖基化水平呈正相关,同时也与tau蛋白的磷酸化水平呈负相关。葡萄糖代谢障碍引起Tau的O-GlcNAC糖基化修饰水平下降,促进了tau的过度磷酸化和AD重要病理改变NFT的形成[8]。
本实验结果显示,大鼠侧脑室注射STZ后第21天即出现明显的学习记忆障碍,模型组与假手术组比较有统计学意义(P<0.01);随着时间的延长第31天后大鼠的学习记忆障碍不但没有恢复,而且有进一步加重趋势(P<0.01)。同时,造模第21天模型组大鼠海马CA1区tau-ps422即有阳性表达,而假手术组则不明显,造模第31天模型组大鼠海马CA1区 tau-ps422表达更为明显,而假手术组几乎没有阳性表达。Western blot检测结果显示:随着时间的延长,造模第31天模型组大鼠在50 kD~71kD之间条带(约67kD,为tau-ps422的表达)明显比假手术组大鼠在这一区域的条带粗,说明造模第31天模型组大鼠海马tau蛋白在Ser422位点的磷酸化水平明显高于假手术组。近几年对于tau蛋白磷酸化位点的研究表明,Ser422位点是AD特异性位点,其磷酸化使tau变成毒性分子从而拮抗正常微管相关蛋白的作用[9],Thr231,Ser396和 Ser422的进一步磷酸化则可促使tau自身聚积成纤维状结构。Tau羧基端(Ser396,Ser404,Ser422)的磷酸化对其形成双股螺旋丝(PHF)起关键作用[10-12]。
STZ侧脑室注射可引起大鼠学习记忆障碍及海马组织tau蛋白AD特异性磷酸化位点Ser422的磷酸化水平增加,这对于进一步形成PHF/NFT非常重要,因而是较为理想的散发性AD(SAD)模型,可用于AD的脑能量代谢及tau蛋白磷酸化方面的研究。
[1] Blokland A,Jolles J.Spatial learning deficit and reduced hippocampal ChAT activity in rats after an ICV injection of streptozotocin[J].Pharmacol Biochem Behav,1993,44:491-494.
[2] Sharma M,Gupta YK.Intracerebroventricular injection of streptozotocin in rats produces both oxidative stress in the brain and cognitive impairment[J].Life Sciences,2000,68(9):1021-1029.
[3] 包新民,舒斯云.大鼠脑立体定向图谱[M].北京:人民卫生出版社,1991:12-18.
[4] Hoyer S,Hennenberg N,Knappet S,et al.Brain glucose metabolism is controlled by amplification and desensitization of the neuronal insulin receptor[J].Ann NY Acad Sci,1996,777:374-379.
[5] Frolich L,Blum-Degen D,Bernstein HG,et al.Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease[J].J Neural T ransm,1998,105(4-5):423-438.
[6] Münch G,Schinzel R,Loske C,et al.Alzheimer's disease-sy nergistic effects of glucose deficit,oxidative stress and advanced glycation endproducts[J].J Neural T ransm,1998,105(4-5):439-461.
[7] Lannert H,Hoyer S.Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats[J].Behav Neurosci,1998,112:119-208.
[8] Liu Y,Liu F,Iqbal K,et al.Decreased glucose transporters correlate to abnormal hyperphos-phory lation of tau in Alzheimer's disease[J].FEBS Lett,2008,582(2):359-364.
[9] Alonso A,Del C,Mederl Yova A,et al.Promotion of hyperphosphorylation by frontotempo-ral dementia taumutations[J].J Biol Chem,2004,279:34878-34881.
[10] Abraha A,Ghoshal N,Gambl In T C,et al.C-terminal inhibition of tau assembly in vitro and in Alzheimer's disease[J].J Cell Sci,2000,113:3737-3745.
[11] Haase C,Stieler J,Arend T T,et al.Pseudophosphorylation of tau p rotein alters its ability for self-aggregation[J].J Neurochem,2004,88:1509-1520.
[12] Ferrar IA,Hoerndl IF,Baech IT,et al.Beta-amyloid induces paired helical filament-like tau filaments in tissue culture[J].J Biol Chem,2003,278:40162-40168.
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国医学影像技术(2020年8期)2020-09-25
天津中医药(2019年9期)2019-09-18
天津医科大学学报(2019年6期)2019-08-13
中国妇幼健康研究(2019年2期)2019-03-26
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
