
普卢利沙星胶囊的含量及有关物质的HPLC测定
2010-06-20 10:50徐耀华肖方青
湖南中医药大学学报 2010年8期
徐耀华,欧 艳,马 宁,肖方青
(1.湖南省第六工程有限公司建设医院,湖南 长沙 410007;2.长沙市中心医院,湖南 长沙 410004;3.湖南医药工业研究所,湖南 长沙 410014)
普卢利沙星(pmlif l oxacin)是一种新的喹诺酮类广谱抗菌药。本品对革兰阴性菌和阳性菌有广谱抗菌作用,特别是对绿脓杆菌为首的革兰氏阴性菌的抗菌力强,可用于全身感染的治疗,其作用优于环丙沙星、氧氟沙星和依诺沙星。本品毒副作用小,口服吸收良好,耐受性好。有关普卢利沙星的测定已有报道[1-3]。我们采用HPLC法,在流动相中引入离子对试剂,测定了普卢利沙星胶囊的含量及有关物质,方法专属性强,重现性好,准确度高,结果满意。
1 仪器与试药
Waters 510泵,2487双波长紫外检测器,HS色谱工作站(杭州英谱科技有限公司)
普卢利沙星对照品(湖南医药工业研究所自制):经结构确证图谱解析与国外文献报道一致,采用HPLC法进行纯度检查,归一法计算含量为99.9%,非水滴定法测定含量为99.9%。乙腈为色谱纯,水为双蒸水,十二烷基硫酸钠为分析纯。普卢利沙星胶囊由湖南医药工业研究所制剂室提供,理论片重为195 mg,规格为132 mg(以C21H20FN3O6S计)。
2 方法与结果
2.1 色谱条件
色谱柱 PICO·TAG TM C18 (5μm,3.9 mm×150 mm ),流动相 乙腈-水-十二烷基硫酸钠(425:500:2.5,用磷酸调节pH为3.0),检测波长 275 nm,流速 1.0 mL/min,柱温 室温,进样量 20 μL。在上述色谱条件下,理论板数按普卢利沙星峰计应不低于2000,普卢利沙星与各有关物质达到基线分离。
2.2 普卢利沙星与合成中间体的分离检测
本品原料药在合成过程中,主要有中间体1:4-乙酰氧基-2-乙硫基-6,7-二氧喹啉-3-羧酸乙酯,2:6,7-二氟-1-甲基-4-氧代-41-1-[1,3]硫氮杂环丁烷并[3,2-a]喹啉-3-羧酸一酯,3:6,7-二氧-1-甲基-4-氧代-41-1-[1,3]硫氮杂环丁烷并[3,2-a]喹啉-3-羧酸。依法配制测定溶液,混合后进样20 μL,记录色谱图1-A。
2.3 辅料的干扰性试验
称取空白辅料9.5 mg,置于100 mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,过滤,取滤液20 μL进样,记录色谱图1-B,同时取胶囊样品细粉适量,依法测定,记录色谱图1-C。
2.4 强制破坏性试验降解产物的检测
取样品适量,分别置于下述条件下,进行强制破坏性试验并对降解产物进行检测。
2.4.1 氧化破坏试验 取普卢利沙星胶囊内容物细粉27.5 mg,置于50 mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,过滤,取滤液4 mL,加双氧水6 mL,放置1.5 min后,进样20 μL,记录色谱图1-D。
2.4.2 碱性破坏试验 取普卢利沙星胶囊内容物细粉30.2 mg,置于50 mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,过滤,取滤液4 mL加 1 moL/L氢氧化钠溶液6 mL,放置40 min,用1 moL/L盐酸溶液调节pH=7,进样20 μL,记录色谱图1-E。
2.4.3 酸性破坏试验 取普卢利沙星胶囊内容物细粉17.6 mg,置于50 mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,过滤,取滤液4 mL加1 moL/L盐酸溶液6 mL,放置1.5 h,用1 moL/L氢氧化钠溶液调节pH=7,进样20 μL,记录色谱图1-F。
2.4.4 光照试验 取普卢利沙星胶囊数粒,置照度为4500 LX的强光下,放置10 d。取出,倾取内容物,研细,称取细粉17.7 mg,置于100 mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,过滤,取滤液20 μL进样,记录色谱图1-G。
通过对普卢利沙星合成中间体,胶囊主药、辅料及降解产物进行分析检测,结果表明采用高效液相色谱法测定普卢利沙星胶囊的含量及有关物质的方法专属性良好。见图1。
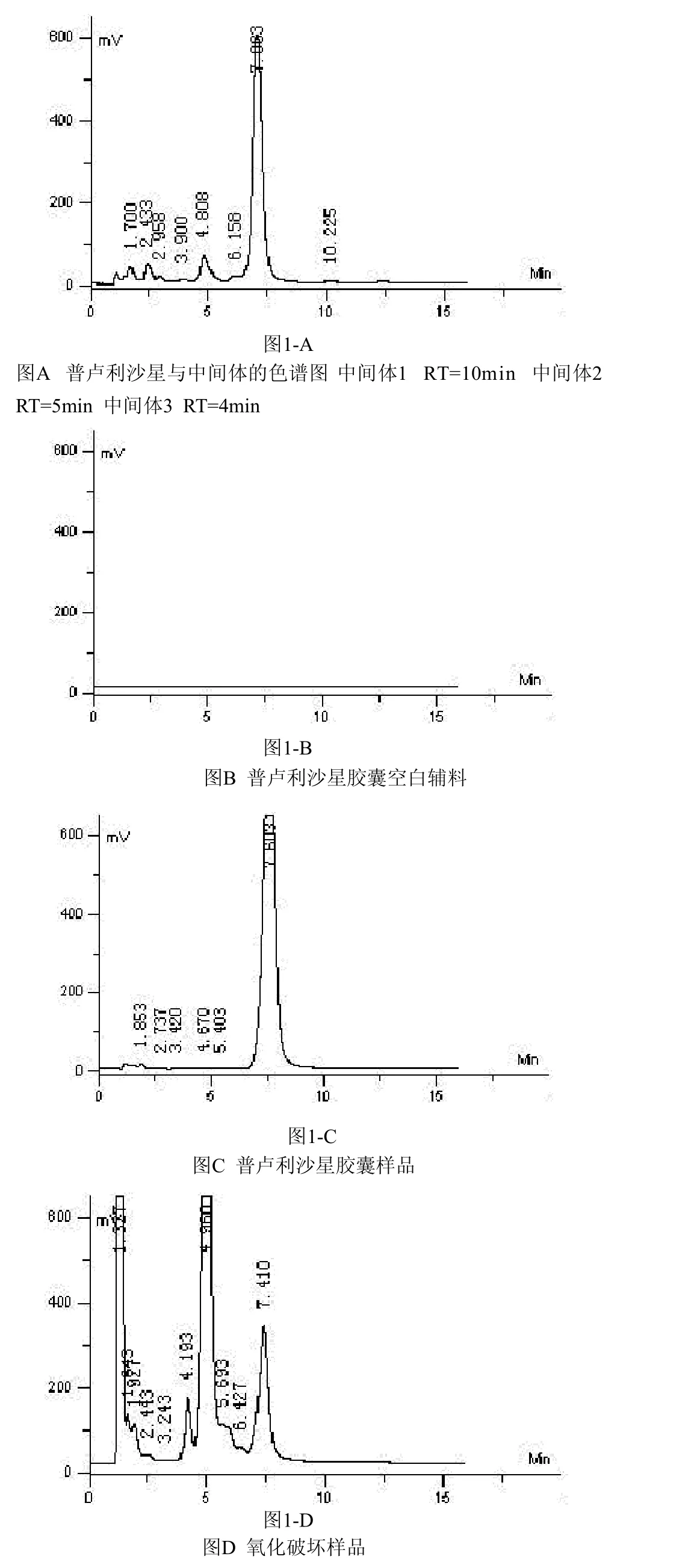
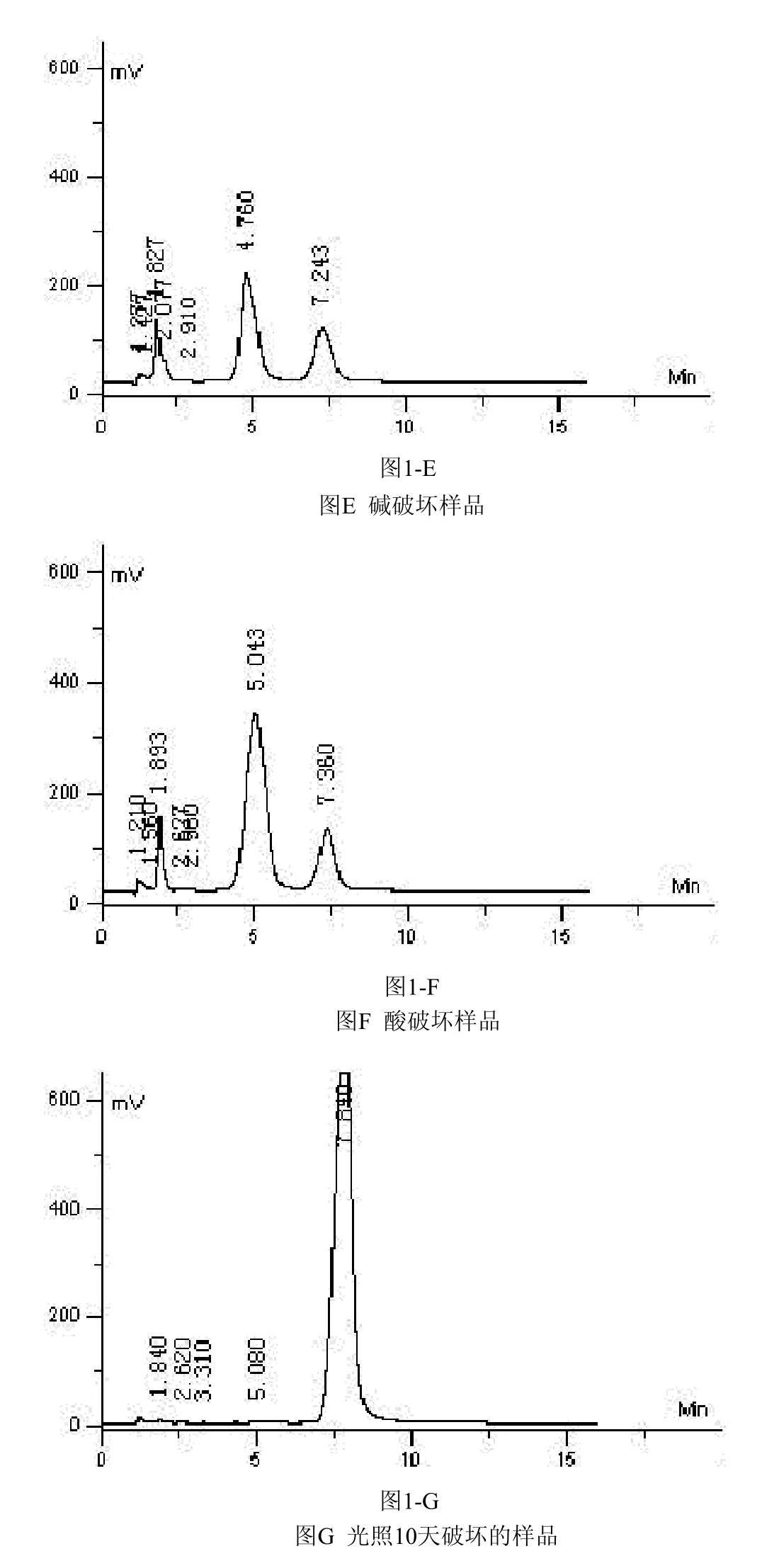
图1 HPLC方法专属性考察图普
2.5 线性试验
取在五氧化二磷干燥剂60℃干燥至恒重的普卢利沙星对照品适量,用乙腈溶解并配成溶液C=0.241 mg/mL的溶液,分别精密吸取5.0,7.5,10.0,12.5,15 mL分别置5个100 mL量瓶中,用乙腈溶液稀释至刻度,摇匀,分别取20 μL注入HPLC仪,记录色谱图。以峰面积S对浓度C进行线性回归,得回归方程为:S=282719+37932C ( r=0.9999 )。表明普卢利沙星在12~36 μg/mL浓度范围内线性关系良好。
2.6 精密度实验和溶液稳定性试验
取“2.5”项下普卢利沙星浓度为25 μg/mL的溶液,连续进样6次,以峰面积考察其精密度,RSD为0.51 % 。
另取“2.5”项下普卢利沙星浓度为25 μg/mL的溶液,分别于室温放置0、2、4、6、8、12、24 h后进样测定,计算得峰面积 RSD=0.41% 。表明室温下溶液在24 h内稳定。
2.7 回收率试验
按普卢利沙星胶囊的处方配比组成制成不含主药的空白辅料,根据胶囊中药物与辅料的比例,在空白辅料中精密加入普卢利沙星对照品,按含量测定方法测定,结果平均回收率为99.7%;RSD分别为0.3%,测定结果符合要求。
2.8 样品测定
含量测定 取装量差异项下普卢利沙星的内容物,混合均匀,精密称取适量(约相当于含普卢利沙星12.5 mg),置于100 mL量瓶中,用乙腈溶液溶解并稀释至刻度,摇匀,滤过,取续滤液5 mL置于25 mL量瓶中,用乙腈稀释至刻度摇匀,作为供试品溶液,取20 μL,注入液相色谱仪,记录色谱图;另取经五氧化二磷60℃减压干燥至恒重的普卢利沙星对照品适量,用乙腈溶解并稀释成每1 mL中约含普卢利沙星25 μg的溶液,同法测定,按外标法以峰面积计算含量。见表1。
有关物质测定 精密称取供试品细粉适量(约相当于普卢利沙星10 mg),置于100 mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,过滤,取续滤液作为供试品溶液。量取1.0 mL置于100 mLl量瓶中,同法稀释至刻度,摇匀,作为对照溶液。量取20 μL注入液相色谱仪,调节检测灵敏度,使普卢利沙星色谱峰的峰高为满量程的15%~30%。再量取供试品溶液和对照溶液各20 μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的2倍。供试品溶液的色谱图中如有杂质峰(除去溶剂峰),各杂质峰面积之和不得大于对照溶液主峰面积(1.0%)。
测定结果 对3批普卢利沙星胶囊分别进行含量测定和有关物质检查,结果见表1。

表1 样品测定结果/%(n=3)
2.10 重复性实验
取同一供试品,分别取样6次,按含量测定项下方法测定含量,结果普卢利沙星的含量RSD为0.25 %,表明重复性良好。
3 讨论
在色谱系统建立过程中,我们参考文献[4]在流动相中引入离子对试剂。在方法学考察中,发现离子对试剂的浓度对色谱峰影响很大。分别取用十二烷基硫酸钠0.5 g,1 g,2 g,2.5 g,3.0 g,3.5 g,配制流动相进行试验,结果表明:(1)十二烷基硫酸钠浓度越高,色谱峰峰形越好,杂质分离度及柱效越高。(2)十二烷基硫酸钠在1 000 mL乙腈-水中溶解度有限,多于3 g开始出现浑浊,而按文献报道[4]十二烷基硫酸钠取用量为3.5 g时,溶液呈半透明的胶体,说明浓度过高,而我们采用在1 000 mL溶剂加2.5 g十二烷基硫酸钠,配成的流动相分离效能高,稳定性良好。
[1]NAKASIIIMA M,UEMATSU T,KQSUGE K,et al.Phammcokinetics and safety of NM441,a new quinolone,in healthy male volunteers [J].J Clin Pharmacol,1994,34(9):930-937.
[2]PIC0LLO R,BRI0N N,GUALANO V,et a1.Pharmacokinetics and tolerability of prulifloxacin after single oral administration [J].Arzneimittelforschung,2003,53(3):20l-205.
[3]封宇飞,李可欣,吕允凤,等,HPLC测定普卢利沙星胶囊的含量和有关物质[J].中国药学杂志,2006,41(2):308-310.
[4]陈玉龙,李立威.加替沙星的HPLC测定[J].中国医药工业杂志,2001,32(12):553.
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
环境卫生工程(2021年3期)2021-07-21
环境卫生工程(2020年3期)2020-07-27
供水技术(2020年6期)2020-03-17
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
山东化工(2018年15期)2018-09-20
数学小灵通(1-2年级)(2017年9期)2017-10-13
学苑创造·B版(2017年1期)2017-02-21
小天使·二年级语数英综合(2016年9期)2016-05-14
环境科技(2015年2期)2015-11-08
