
Synthesis, characterization, DNA-binding of complexes [Ru(phen)2(7-R-dppz)]2+( R = -CH3, -F, -CF3)
2010-05-09 07:52LIUPingLIUXuewenLUJilinCHENYuandaoLILin
湖南文理学院学报(自然科学版) 2010年3期
LIU Ping, LIU Xue-wen ,2, LU Ji-lin , CHEN Yuan-dao ,2, LI Lin
Synthesis, characterization, DNA-binding of complexes [Ru(phen)2(7-R-dppz)]2+( R = -CH3, -F, -CF3)
LIU Ping1, LIU Xue-wen1,2, LU Ji-lin1, CHEN Yuan-dao1,2, LI Lin1
(1. College of Chemistry and Chemical Engineering, Hunan University of Arts and Science, Changde 415000, China;2. Key Lab of Environment-Friendly Chemistry and Application in Ministry of Education, Xiangtan University, Xiangtan 411105, China)
Three Ru(II) polypyridyl complexes, [Ru(phen)2(7-CH3-dppz)]2+(i), [Ru(phen)2(7-F-dppz)]2+(ii) and [Ru(phen)2(7-CF3-dppz)]2+(iii) have been synthesized and characterized by elemental analysis, ES-MS (electrospray mass spectra),1HNMR. The DNA-binding behaviors of these complexes have been studied by spectroscopic methods and viscosity measurements. The results indicate that the three complexes all bind to CT-DNA (calf thymus DNA) in an intercalative mode, and the DNA-binding affinity follows the order of (ii) > (i) > (iii). Theoretical studies for these complexes have been also carried out with the density functional theory (DFT) method. The difference in the DNA-binding behaviors of three complexes can be reasonably explained by the DFT calculations.
Ru(II) complex; polypyridyl ligand; DNA-binding; DFT.
During the past decades, Ru(II) polypyridyl complexes have been widely studied due to their pote- ntial applications in stereoselective probes of nucleic acid structures, molecular “light switches”, DNA- photocleavage and optical reagents[1-9]. It is well- established that these Ru(II) complexes can bind to DNA in a noncovalent interaction such as electrostatic binding, groove binding, or intercalation mode, in which the intercalation mode is the most interesting one because there is strong π–π stacking interaction between the intercalative ligand of the complex and DNA-base-pairs. Such an interaction not only closely relates to their above-mentioned wide applications, but also offers a wide and interesting research field for experimental and theoretical works[3, 10-15].
The Ru(II) polypyridyl complexes with great potential applications have inspired vigorous interests of scientists to continuously design novel species. A great number of reports have shown that via impro- ving the structure of ligand, in particular, intercalative ligand, most of resulting Ru(II) polypyridyl-type com- plexes were found to have special DNA-binding, DNA-photocleavage and spectral properties so that their applications in biochemistry and photochemistry were further developed[3]. Dipyridophenazine (dppz) complexes of Ru (II) provides sensitive luminescent probes for double stranded DNA in soultion. [Ru(L)2(dppz)]2+(L=bpy, phen) show no photolum- inescence in aqueous solution at ambient temperature but luminesce brightly upon binding intercalatively with the dppz ligand between adjacent base pairs, and exhibit much stronger DNA-binding affinity ([Ru(phen)2(dppz)]2+(= 5.1 × 106M-1 [16])). How- ever, varying the nature or location of the substituent in the intercalative ligand can create some interesting differences in the space configuration and electron density distribution of Ru(II) polypyridyl complexes, resulting in differences in spectral properties, DNA- binding behaviors, and even photocleavage properties.
In order to obtain more insights into the DNA- binding properties of Ru(II) complexes, a series of derivatives of [Ru(phen)2(dppz)]2+, [Ru(phen)2(7-R- dppz)]2+(R=-CH3, -F, -CF3) have been synthesized and characterized. We expect these novel Ru(II) complexes have some improved properties in DNA- binding, and spectral behaviors by introducing electron-withdrawing or electron-pushing substituents on the typical intercalative ligand, dppz. Therefore, in this paper, the DNA-binding and spectral properties of these novel Ru(II) complexes were carefully studied. In addition to experimental works, the theoretical study of these complexes using DFT methods was also carried out in order to reveal the trend in their DNA binding affinities as well as explain their spectral properties.
1 Experimental
All reagents and solvents were purchased commercially and used without further purification unless otherwise noted. The dialysis membrane was purchased from Union Carbide Co. And treated by means of general procedure before use[17]. Solutions of CT-DNA (calf thymus DNA) in 50 mM NaCl, 5 mM Tris–HCl(tris(hydroxymethyl)aminomethane hydroch- loride) (pH=7.2) gave a ratio of UV-Vis absorbance of 1.8~1.9:1 at 260 and 280 nm, indicating that the DNA was sufficiently free of protein[17]. The concentration of DNA was determined spectrophotometrically using a molar absorptivity of 6 600 M−1﹒cm-1(260 nm)[18]. Double-distilled water was used to prepare buffers. The compounds-[Ru(phen)2Cl2]﹒2H2O[19]and 1, 10-phenanthroline-5, 6-dione(phendione)[20]were pre- pared by the literature methods.
1.1 Physical measurement
Microanalyses (C, H, and N) were carried out with a Perkin–Elmer 240Q elemental analyser. Fast atom bombardment (FAB) mass spectra were measured on a VG ZAB-HS mass spectrometer with 3-nitrobenzyl alcohol as matrix and electrospray mass spectra (ES-MS) were recorded on a LQC system (Finngan MAT, USA) using CH3CN as mobile phase. The spray voltage, tube lens offset, capillary voltage and capillary temperature were set at 4. 50 kV, 30.00 V, 23.00 V and 200°C, respectively, and thevalues were quoted for the major peaks in the isotope distribution.1H NMR spectra were recorded on a Bruker ARX-500 spectrometer with (CD3)2SO or CDCl3as solvent for the ligands and CD3CN or (CD3)2SO for the complexes at 500 MHz at room temperature. All chemical shifts relative to TMS (tet- ramethylsilane) were given. UV-Vis (UV-visible) spectra were recorded on a Perkin Elmer Lambda850 spectrophotometer and emission spectra were recorded on a Perkin Elmer Ls55 spectrofluorophoto meter at room temperature.
The absorption titrations of Ru(II) complexes in buffer(5 mM Tris-HCl (tris(hydroxymethyl) aminome- thane hydrochloride), 50 mM NaCl, pH=7.2) were performed by using a fixed ruthenium concentration to which increments of the DNA stock solution were added. Ruthenium solutions employed were 20mM in concentration. Ruthenium-DNA solutions were allow- ed to incubate for 10 min before the absorption spectra were recorded.
Viscosity measurements were carried out using an Ubbelohde viscometer maintained at a constant temperature of 30.0±0.1°C (in a thermostatic bath). Flow time was measured with a digital stopwatch and every sample was measured three times and an average flow time was calculated. Data were presented as (/0)1/3binding ratio[21], whereis the viscosity of DNA in the presence of complex and0is the viscosity of DNA alone.
Equilibrium dialysis measurements were conduc- ted at room temperature with 5 cm3of CT-DNA (0.5 mM) sealed in a dialysis bag (cellulose membrane) and 10 cm3of the complexes (20mM) outside the bag with the solution stirred for 48 h. In the control exp- eriments, 5 cm3Tris-HCl buffer was used instead of CT-DNA. Before use, the dialysis membranes were boiled for1 h in a 1% EDTA (ethylenediaminet- etraacetic acid) and 3% NaHCO3solution and then rinsed in doubly deionized water.
1.2 Synthesis
1.2.1 7-CH3-dppz (1a)[22]
A solution of phendione (0.210 g, 1 mmol) and 4-methyl-1,2-phenylenediamine (0.122 g, 1 mmol) in ethanol (20 ml) was heated at reflux for 4 h. After cooling, the yellow precipitate was collected by filtration, washed with cold ethanol, and vacuum-dried. Yield: 0.214 g, 78.4% (Found: C, 77.04; H, 4.11; N, 18.89%. Calcd for C19H12N4: C, 77.01; H, 4.08; N 18.91%. ); FAB-MS:297 ([M+1]+)
1.2.2 7-F-dppz (2a)
This pale white compound was prepared by a similar procedure described for 7-CH3-dppz, 4-F-1, 2-phenylenediamine (0.126 g, 1 mmol) was used instead of 2,3-diaminophenol. Yield: 0.218 g, 60.8%. (Found: C, 72.02; H, 3.05; N, 18.65%. Calcd for C18H9N4F: C, 72.00; H, 3.02; N 18.66%. ); FAB-MS:301 ([M+1]+);1H NMR (500 Hz, ppm, CDCl3): 9.58(dd, 2H), 9.32(m, 2H), 8.34(dd, 1H), 8.34(d, 1H,=9.0 Hz), 7.79(dd, 1H), 7.71(m, 2H).
1.2.3 7-CF3-dppz(3a)
This pale white compound was prepared by a similar procedure described for 7-CH3-dppz, 4-CF3-1, 2-phenylenediamine (0.176 g, 1 mmol) was used instead of 2, 3-diaminophenol. Yield: 0.218 g, 55.2%. (Found: C, 65.17; H, 2.61; N, 15.94%. Calcd for C19H9N4F3: C, 65. 15; H, 2.59; N 15. 99%. ); FAB-MS:351 ([M+1]+);1H NMR (500 Hz, ppm, CDCl3): 9.62(m, 2H), 9.32(m, 2H), 8.66(s, 1H), 8.46(d, 1H,=9.0 Hz), 8.08(dd, 1H), 7.83(dd, 2H).
1.2.4 [Ru(phen)2(7-CH3-dppz)](ClO4)2(i)
A mixture of-[Ru(phen)2Cl2]. 2H2O (0.114 g, 0.2 mmol), 7-CH3-dppz (0.060 g, 0.2 mmol) and glycol(20 mL) was refluxed under argon for 2 h. Upon cooling, the resulting clear red solution was diluted with 60 mL water. After filtration, the solution was treated with a saturated aqueous solution of NaClO4, and a red precipitate was obtained. The crude product was purified by column chromato- graphy on alumina with acetonitrile- toluene (1:1,/) as an eluent. The red band was collected. The solvent was removed under reduced pressure and red microcrystals were obtained. Yield: 0.125 g, 64% (Found: C, 53.86; H, 3.01; N, 11.68%. Calcd for C43H28N8O8RuCl2: C, 53.97; H, 2.95; N, 11.72%); ES-MS (CH3CN):= 857.0 ([M2++ClO4]+), 379.3 (M2+);1H NMR (500 MHz, ppm, DMSO-6): 9.61 (dd, 2H), 8.79 (dd, 4H), 8.41 (t, 1H), 8.39 (s, 4H), 8.29 (m, 3H), 8.16(td, 2H), 8.05 (dt, 3H), 7.91 (m, 2H), 7.77 (m, 4H), 2.74 (s, 3H)
1.2.5 [Ru(phen)2(7-F-dppz)](ClO4)2(ii)
This complex was obtained by a similar procedure described for complex (i). 7-F-dppz (0.070 mg, 0.2 mmol) was used instead of 7-CH3-dppz. The crude product was purified by column chromato- graphy on alumina with acetonitrile-toluene (2:1,/) as an eluent. The red band was collected. The solvent was removed under reduced pressure and red micro- crystals were obtained. Yield: 0.131 g, 64% (Found: C, 52.48; H, 2.69; N, 11.62%. Calcd for C42H25N8F- O8RuCl2: C, 52.50; H, 2.62; N, 11.67%); ES-MS (CH3CN):= 861.1 ([M2++ClO4]+), 381.3 (M2+);1H NMR (500 MHz, ppm, CD3CN): 9.62 (d, 2H,= 8.5 Hz), 8.63 (t, 4H), 8.53 (dd, 1H), 8.28 (s, 4H), 8.22 (tt, 2H), 8.28 (m, 3H), 8.02 (d, 2H), 7.99 (t, 1H), 7.79 (dd, 2H), 7.66 (m, 4H)
1.2.6 [Ru(phen)2(7-CF3-dppz)](ClO4)2(iii)
This complex was obtained by a similar procedure described for complex 1.7-CF3-dppz (0. 065 mg, 0.2 mmol) was used instead of 7-CH3-dppz. The crude product was purified by column chromatogra- phy on alumina with acetonitrile-toluene (3:1,/) as an eluent. The red band was collected. The solvent was removed under reduced pressure and red micro- crystals were obtained. Yield: 0.131 g, 64% (Found: C, 50.99; H, 2.48; N, 11.04%. Calcd for C43H25N8F3O8- RuCl2: C, 51.09; H, 2.49; N, 11.09%); ES-MS (CH3CN):= 912. 9 ([M2++ClO4]+), 406.3(M2+);1H NMR (500 MHz, ppm, DMSO-6): 9. 61 (dd, 2H), 8. 90 (s, 1H), 8. 79 (t, 4H), 8. 73 (d, 1H,= 9. 0 Hz), 8. 40 (s, 4H), 8. 28 (m, 3H), 8. 20 (d, 2H,= 4. 5 Hz), 8. 05 (d, 2H,= 9. 0 Hz), 7. 94 (dd, 2H), 7. 78 (m, 4H).CAUTION! Perchlorate salts of metal complexes with organic ligands are potentially explosive, and only small amounts of the material should be prepared and handled with great care.
1.2.7 Theoretical Section
The structural schematic diagrams of these complexes are shown in Fig 1. Each octahedral complex of the general structrure [Ru(phen)2L]2+(where L=7-CH3-dppz, 7-F-dppz or 7-CF3-dppz) for- ms from a Ru(II) ion, a main intercalative ligand (L), and two phen co-ligands. There is not any symmetry in these complexes. All calculations have been perfor- med with the Gaussian 98 quantum chemistry prog- ram-package[23-24], at the DFT/B3LYP level using 6-31G* basis set on the carbon, nitrogen, fluorine and hydrogen atoms and a LanL2DZ pseudo potential[25-26]on the ruthenium atom. The full geometry optimiz- ation computations for the ground states of these complexes with singlet state[27]were carried out. In order to vividly depict the detail of the frontier molecular orbital interactions, the stereographs of some related frontier molecular orbitals of the complexes were drawn with the Molden v3.7 progr- am[28]based on the computational results.

Fig 1 Structural schematic diagrams of [Ru(phen)2(7-R-dppz)]2+(R =-CH3,-F,-CF3)
2 Results and discussion
2.1 Synthesis and characterization
The ligands was prepared by a method similar to that described by A. Arancibia et.al[22]. These comple- xes were prepared by direct reaction of 7-R-dppz with the equal mole ratios of the precursor complexes [Ru(phen)2Cl2]. The yields were moderate in each case. The desired Ru(II) complexes were isolated as the perchlorates and purified by column chromatography. In the ES-MS spectra for the complexes, only the signals of [M-ClO4-]+and [M-2ClO4-]2+were observed. The measured molecular weights were cons- istent with expected values.
2.2 Electronic absorption titration
The electronic absorption spectra of these Ru (II) complexes are shown characterized by intense π-π*ligand transitions in the UV region and MLCT transition in the visible region. The bands below 300 nm are atributed to intraligand (IL) π-π*transitions, the bands of the complexes (i), (ii), (iii) at 377, 375 or 370 nm are atributed to the π-π*transitions, and the lower energy bands at 438 nm are assigned to the metal-to-ligand charge transfer transitions by comparsion with the spectra of other polypyridyl Ru(II) complexes.
The absorption spectra of complex (i), (ii), (iii) in the absence and presence of CT-DNA (at a constant concentration of complexes, [Ru] = 20 μM) are shown in Fig 2. As the concentration of DNA is increased, the MLCT transitions bands of complex (i), (ii), (iii), at 438 nm exhibt hypochromism of about 17. 8%, 16. 8% and 18.5%, and bathochromism of 2 nm for complex (i) and (ii), respectively. No obvious red shift is observed for complex (iii). The spectroscopic changes suggest that there exist some interactions between these Ru(II) complexes and DNA.
In order to compare quantitatively the DNA- binding affinities of the three complexes, the intrinsic binding constantsof the complexes to DNA were obtained by monitoring the changes of the MLCT absorbance at 438 nm for complexes according to eq. 1[29-31].

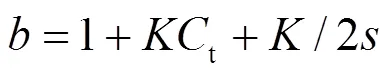
whereais the extinction coefficient observed for the MLCT absorption band at a given DNA concentration,fis the extinction coefficient of the complex in the absence of DNA,bis the extinction coefficient of the complex fully bound to DNA (when further addition of DNA does not change the absorbance, it is assumed that all complex specimen has been bound to DNA andbcan be calculated from Beer’s Law).is the equilibrium binding constant in M-1,tis the total metal complex concentration, [DNA] is the concent- ration of DNA in M (nucleotide), andis the binding size. The experimental absorption titration data were fitted to obtain the binding constants by a non-linear least-square method. In plots of (a-f)/ (b-f) vs. [DNA],was given. The intrinsic binding constants of complex (i), (ii), and (iii) were 1.25 × 106M-1, 2.69×106M-1, and 8. 6×105M-1, respectively. The values are smaller as expected than that of their parent complex [Ru(phen)2(dppz)]2+(=5.1×106M-1)[16]. The results show that the DNA affinity of these complexes closely correlate to the electronic effects and the steric hindrance of the substituent group of their intercala- ting ligand. As reported before, the DNA-binding affinity of complexes can be influenced by electronic effects of intercalative ligands[32-33]. Here complex (ii) exhibits a stronger DNA-binding affinity than (i) and (iii). This result shows that the electron-withdrawing substituent(-F) on the intercalative ligand can increase the DNA-binding affinity of the complex, whereas the electron-donating substituent (-CH3) has the opposite effect. The lowest DNA affinity of complex (iii) is attributed to the lagerest steric hindrance of the electron-withdrawing substituent (-CF3). This trend suggests us that the DNA-binding affinity of the complex can be effectively cotrolled by the substituents.

Arrow shows the absorbance changing upon the increase in DNA concentration. Inset: plots of (a-f)/(b-f) vs. [DNA] for the titration of DNA to Ru(II) complexes.
Fig 2 Absorption spectra of complexes (i)(a) ,(ii)(b), (iii)(c) in Tris-HCl buffer upon the addition of CT-DNA, [Ru] = 20 μM, [DNA] = 0 ~ 350 μM.
2.3 Emission spectra
In the absence of DNA, complex (i), (ii) and(iii) can emit weak luminescence in Tris buffer at ambient temperature, with maximum at ca. 599, 592 and 593 nm, respectively. Upon addition of CT-DNA, the emission intensity of these complexes increase steadily to255.8, 26.3 and 5.0 times of the original, respectively (Fig 3). This result implies that these complex can interact with CT DNA and be protected by DNA efficiently, since the hydrophobic environ- ment inside the DNA helix reduces the accessibility of solvent water molecules to the complex and the complex mobility is restricted at the binding site, leading to decrease of the vibrational modes of relaxation.
2.4 Viscosity properties
The viscosity experiments were performed to further clarify the nature of interaction between DNA and the complex. Optical photophysical probes provide necessary but not sufficient clues to support the binding mode. Hydrodynamic measurements being sensitive to length change (i.e. viscosity and sedimen- tation) are regarded as the least ambiguous and most critical tests for a classical intercalation model in solution in the absence of crystallographic structural data[34-35]. A classical intercalation model demands that the DNA helix lengthens as base pairs are separated to accommodate the bound ligand, leading to the increase of DNA viscosity. In contrast, a partial, non-classical intercalation of ligand could bend (or kink) the DNA helix, reducing its length and, concomitantly, its viscosity[34-35].
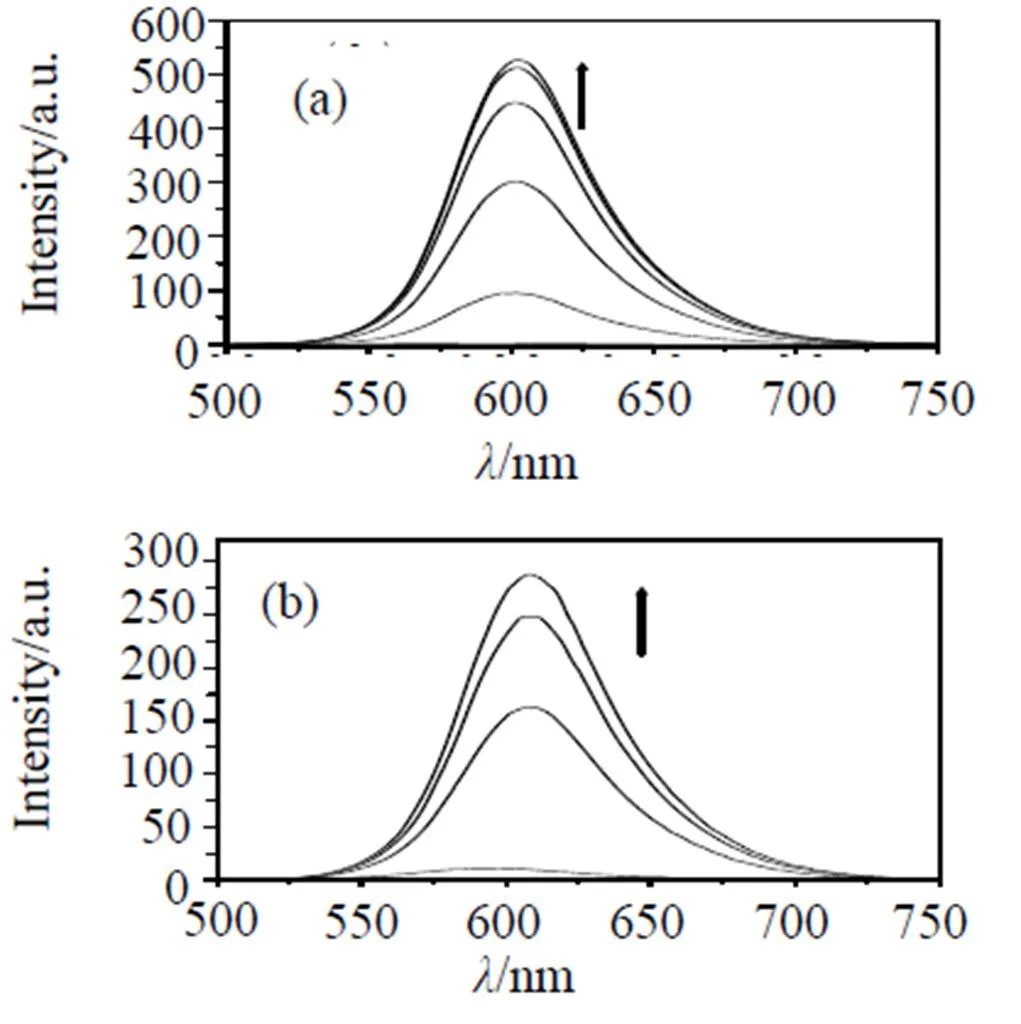
Arrow shows the intensity change upon the increase in DNA concentration. Samples were excited at 438 nm, respectively; emission spectra were monitored between 500 and 800 nm.
Fig 3 Emission spectra of Ru(II) complexes (i)(a) ,(ii)(b), (iii)(c) (2 μM) in Tris-HCl buffer at 298 K in the absence and the presence of calf thymus DNA.
The effects of complex (i), (ii), (iii) and [Ru(bpy)3]2+(bpy=2,2¢-bipyridine) on the viscosity of rod-like DNA are shown in Fig 4. For complex [Ru(bpy)3]2+, which has been known to bind to DNA in an electrostatic mode, it exerts essentially no effect on DNA viscosity. On increasing amount of comple- xes, the relative viscosity of DNA increases steadily. The extent of increase in viscosity, which may depend on the DNA-binding affinity, follows the order of (ii)>(i)>(iii)>[Ru(bpy)3]2+. The results suggest that these complexes intercalate between the base pairs of DNA, which are consistent with the spectroscopic results above.
Fig 4 Effects of the increase in amounts of complexes (i) (▲), (ii)(∆), (iii)(●) and [Ru(bpy)3]2+(■) on the relative viscosity of calf thymus DNA at 30 (±0. 1)°C, respectively. The total concentration of DNA is 0. 25 mM.
2. 5 Enantioselective binding
The equilibrium dialysis experiment may be one of the most direct means of examining the enantiosel- ectivity of the complex binding to DNA. According to the DNA-binding model, the Δ enantiomer binds preferentially to the right-handed helix[36]. The cir- cular dichroism spectra in UV region of complex (i), (ii)and (iii) after their racemic solutions have been dialyzed against CT-DNA for 48 h, are shown in Fig 5. The strong CD signals appear for complex (i), (ii), (iii) with a negative peak at 252, 254 and 255 nm, respectively and a positive peak at 268 nm. Although neither of the complexes have been resolved into their pure enantiomers, and we cannot determine which enantiomer binds DNA enantioselectively for these complexes, it is certain that complex (i), (ii) and (iii) interact with CT-DNA enantioselectivly.

Fig 5 CD spectra of the dialyzates of complex (i)(solid line), (ii)(dot line) and (iii)(dash line) in the presence of CT- DNA after 48 h dialysis with the stirred solution, [Ru] = 20 μM, [DNA] = 0. 5 mM.
2.6 Theoretical explanation on the electro- nic effects of complexes binding to DNA
The trend in DNA-binding affinities of the complexes can be theoretically explained. The calcu- lated selected geometric data, some frontier molecular orbital energies and molecular orbital stereographs of complex (i), (ii), (iii) are given in Table 1, Table 2 and Fig 6, respectively.
As well-established, there arep-pstacking inter- actions in the DNA-binding of these complexes in an intercalative mode, and the base pairs of DNA are electron-donors[37-38]and the intercalated complex is electron-acceptor. Therefore, based on the frontier molecular orbital theory[39-41], the DNA-binding affinity of the complex should rely on three factors: the first one is the LUMO (the lowest unoccupied molecular orbital) energy of the complex since lower LUMO energy of the complex is advantageous to accepting the electrons offered from base pairs of DNA. The second one is the planarity area of the intercalative ligand because the larger planarity area is advantageous to the interaction between DNA and the complex. The third one is the populations of LUMO and/or some virtual orbitals near LUMO on the intercalative ligand because more such populations are also advantageous to the overlap between HOMO of DNA and LUMO of the complex and thus to the interaction between DNA and the complex.
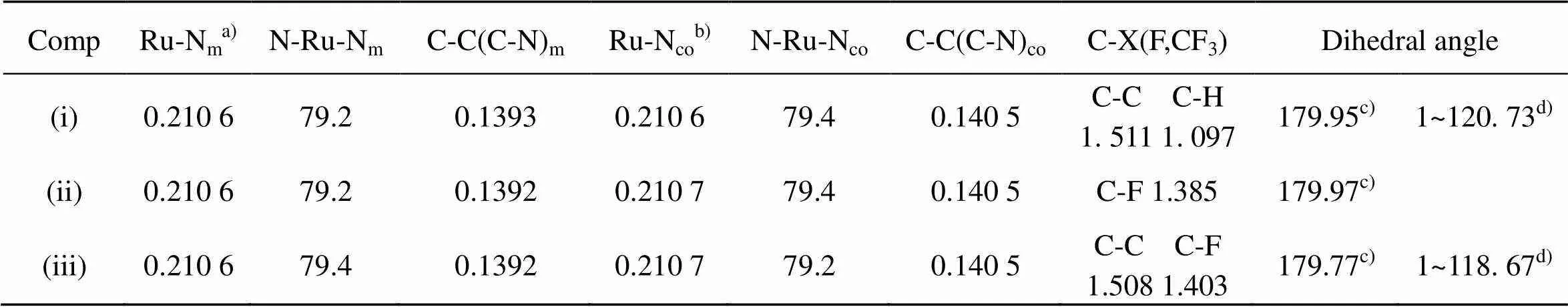
TABLE 1 Calculated selected bond lengths [nm], bond angles and dihedral angles [˚] of (i), (ii) and(iii)
a) Ru-Nm: mean bond length between Ru and the coordinating N-atoms of the main ligand.
b) Ru-Nco: mean bond length between Ru and the coordinating N-atoms of the co-ligand.
c) C(1)-C(2)-C(3)-C(4).
d) F(5)-C(4)-C(2)-C(1).
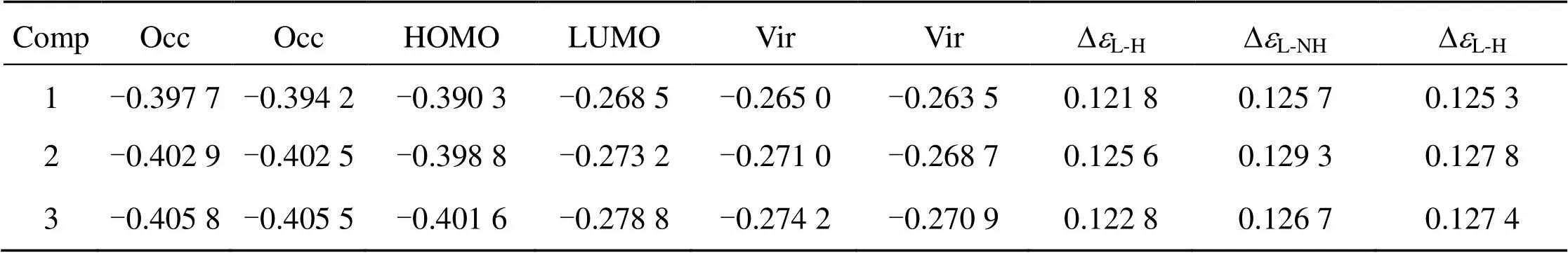
TABLE 2 Some frontier molecular orbital energies (ei/a. u. ) of the complexes (1 a. u. =27. 21 eV).
a) HOMO (or H): the highest occupied molecular orbital.
b) LUMO (or L): the lowest unoccupied molecular orbital.
c)DL-H: the energy difference between LUMO and HOMO;DL-NH: the energy difference between LUMO and NHOMO.
From Table 1 and Fig 6, we can see that the order of the energies () of the lowest unoccupied molecular orbital (LUMO) isLUMO(iii) <LUMO(ii) <LUMO(i), moreover, the LUMO component of (ii) and (iii) is predominantly distributed on the intercalative ligand whereas that of (i) is predominantly distributed on the co-ligands. These two factors should be advantageous to the DNA-binding affinity of complex (ii) and (iii), espectially complex (iii). However, the experiment results show that complex (iii) has the lowest DNA- binding affinity. Therefore, the DNA-binding affinity of the complexes must be considered simultaneously the effects from the both factors, the electronic effect and steric influence of the substituent. Though Com- plex (iii) possesses the strongest electron withdrawing substituent (-CF3), its largest steric hindrance results in the lowest DNA-binding affinity.

Fig 6 Stereographs of HOMOs and LUMOs of [Ru(phen)2(7-R-dppz)]2+(R = -CH3, -F, -CF3).
3 Conclusion
Three Ru(II) complexes [Ru(phen)2(7-R-dppz)]2+(R=-CH3,-F,-CF3) have been synthesized and char- acterized. The DNA-binding properties of these com- plexes have been investigated. The results show that these complexes can bind to DNA in an intercalative mode, and that the electron-withdrawing substituent on the intercalative ligand has a significant effect on DNA-binding affinity (), resulting in(i) <(ii). meanwhile, the steric influence of the substituent also affects the DNA-binding affinity, resulting in the lowest DNA-binding affinity. Synthetically consid- ering these factors, it can reasonably explain above experimental results, in which the DNA-binding constants () are 1.52, 2. 69 and 0. 86 (×106M-1) for complex (i), (ii) and (iii), respectively.
[1] Pyle A, Barton J K, Lippard S J. Progress in Inorganic Chemistry[J]. New York, 1990, 38: 413–475.
[2] Sigel A, Sigel H. MetalIons in Biological Systems[J]. Marcel Dekker, 1996, 33: 177–252.
[3] Ji L N, Zou X H, Liu J G. Shape and enantioselective interaction of Ru(II)/Co(III) polypyridyl complexes with DNA [J]. Coord Chem Rev, 2001, 216-217: 513-536.
[4] Zeglis B M, Pierre V C, Barton J K. Metallointercalators and Metalloinsertors[J]. Chem Commun, 2007, 4565- 4579.
[5] Clarke M J. Ruthenium Metallopharmaceuticals[J]. Coord Chem Rev, 2003, 236: 209-233.
[6] Nazeeruddin M K, Klein C, Liska P, et al. Synthesis of novel ruthenium sensitizers and their application in dye-sensitized solar cells [J]. Coord Chem Rev, 2005, 249: 1460-1467.
[7] Metcalfe C, Thomas J A. Kinetically inert transition metal complexes that reversibly bind to DNA [J]. Chem Soc Rev, 2003, 32: 214-224.
[8] O’Donoghue K A, Kelly J M, Kruger P E. Unusual photophysical switching in a Ru(II) diimine DNA probe caused by amide functionalisation [J]. Dalton Trans 2004, 13-15.
[9] Shi S, Liu J, Li J, et al. Electronic effect of the different positions of-NO2group on the DNA-intercalator of chrial complexes [Ru(bpy)2(L)]2+(L= o-npip, m-npip and p-npip) [J]. Dalton Trans, 2005: 2038-2046.
[10] Li J, Xu L C, Chen J C, et al. Density Functional Theory/ Time-dependent DFT Studies on the Structures, Trend in DNA-binding Affinities, and Spectral Properties of Complexes [Ru(bpy)2(p-R-pip)]2+(R=-OH,-CH3,-H,-NO2) [J]. J Phys Chem A, 2006, 110: 8174-8180.
[11] Nordell P, Lincoln P. Mechanism of DNA threading inter- calation of binuclear Ru complexes: unior bimolecular pathways depending on ligand structure and binding density [J]. J Am Chem Soc, 2005, 127: 9670-9671.
[12] Xu H, Zheng K C, Chen Y, et al. Effects of ligand planarity on the interaction of polypyridyl Ru(II) com- plexes with DNA[J]. Dalton Trans, 2003: 2260-2268.
[13] Xu L C, Shi S, Li J, et al. A combined computational and experimental study on DNA-photocleavage of Ru(II) polypyridyl complexes [Ru(bpy)2(L)]2+(L = pip, o-mopip and p-mopip) [J]. Dalton Trans, 2008: 291-301.
[14] Tan L F, Wang F, Chao H, et al. Ruthenium(II) mixed- ligand complex containing 2-(4′-benzyloxy-phenyl)imid- azo[4,5-f][1,10]phenanthroline: Synthesis, DNA-binding and photocleavage studies [J]. J Inorg Biochem, 2007, 101: 700-708.
[15] Roy M, Pathak B, Patra A K, et al. New Insights into the Visible-Light-Induced DNA Cleavage Activity of Dipyri- doquinoxaline Complexes of Bivalent 3d-Metal Ions[J]. Inorg Chem, 2007, 46: 1112-11132.
[16] Rajesh B N, Emily S T, Shalawn L K, et al. Synthesis and DNA-Binding Properties of [Ru(NH3)4dppz]2+[J]. Inorg. Chem. 1998, 37: 139-141.
[17] Marmur J. A procedure for the isolation of deoxyri- bonucleic acid from micro-organisms[J]. J Mol Biol, 1961, 3: 208-218.
[18] Reichmann M E, Rice S A, Thomas C A, et al. A Further Examination of the Molecular Weight and Size of Desoxypentose Nucleic Acid[J]. J Am Chem Soc, 1954, 76: 3047-3053.
[19] Sullivan B P, Salmon D J, Meyer T J. Mixed phosphine 2,2'-bipyridine complexes of ruthenium[J]. Inorg Chem, 1978, 17: 3334-3341.
[20] Yamada M, Tanaka Y, Yoshimato Y, et al. Synthesis and Properties of Diamino-Substituted Dipyrido [3,2-a: 2′,3′-c] phenazine [J]. Bull Chem Soc Jpn, 1992, 65: 1006-1011.
[21] Cohen G, Eisenberg H. Viscosity and sedimentation study of sonicated DNA–proflavine complexes[J]. Biopolymers, 1969, 8: 45-55.
[22] Arancibia A, Concepcion J, Daire N, et al. Electronic effects of donor and acceptor substituents on dipyrido (3,2-a:2′,3′-c) phenazine(dppz) [J]. J Coord Chem, 2001, 54: 323-336.
[23] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 98[M]. Revision A.11.4. Pittsburgh: PA Gaussian, Inc, 2002.
[24] Foresman J B, Frisch E. Exploring Chemistry with Electronic Structure Methods[M]. 2nd ed. Pittsburgh PA: Gaussian Inc, 1996.
[25] Hay P J, Wadt W R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg [J]. J Chem Phys, 1985, 82: 270-283.
[26] Wadt W R, Hay P J. Ab initio effective core potentials for molecular calculations. Potentials for main group elem- ents Na to Bi [J]. J Chem Phys, 1985, 82: 284-298.
[27] Juris A, Balzani V, Barigelletti F, et al. Ru(II) polypy- ridine complexes: photophysics, photochemistry, eletro- chemistry, and chemiluminescence [J]. Coord Chem Rev, 1988, 84: 85-277.
[28] Schaftenaar G, Noordik J H. MOLDEN: a pre- and post- processing program for molecular and electronic structu- res[J]. J Comput-Aided Mol Design, 2000, 14: 123-134.
[29] Barton J K, Dannenberg J J, Raphael A L. Tris (phenant- hroline) ruthenium(II): stereoselectivity in binding to DNA [J]. J Am Chem Soc, 1984, 106: 2172-2176.
[30] Smith S R, Neyhart G A, Karlsbeck W A. Thorp H.H [J]. New J Chem, 1994, 18: 397.
[31] Carter M T, Rodrigues M, Bard A J. Voltammetric studies of the interaction of metal chelates with DNA. 2. Tris-chelated complexes of cobalt(III) and iron(II) with 1,10-phenanthroline and 2,2'-bipyridine[J]. J. Am. Chem. Soc. 1989, 111: 8901-8911.
[32] Liu J, Mei W J, Lin L J, et al. Electronic effects on the interactions of complexes [Ru(phen)2(p-L)]2+(L=MOPIP, HPIP, and NPIP) with DNA [J]. Inorg Chim Acta, 2004, 357: 285-293.
[33] Mei W J, Ma Yu Z, Liu J, et al. Experimental and DFT Studies on the DNA-binding Properties of Ruthenium(II) Complexes [Ru(phen)2L]2+(L = o-MOP, o-MP, o-CP and o-NP) [J]. Transit Metal Chem, 2006, 31: 277-285.
[34] Satyanarayana S, Dabroniak J C, Chaires J B. Neither DELTA.-or .LAMBDA.-tris(phenanthroline)ruthenium(II)bin-ds to DNA by classical intercalation [J]. Bio- chemistry, 1992, 31: 9319-9324.
[35] Satyanaryana S, Daborusak J C, Chaires J B. Tris (phen- anthroline) ruthenium(II) enantiomer interactions with DNA: Mode and specificity of binding [J]. Biochemistry, 1993, 32: 2573-2584.
[36] Barton J K. Metals and DNA: Molecular Left-Handed Complements [J]. Science, 1986, 233: 727-734.
[37] Reha D, Kabelac M, Ryjacek F, et al. Intercalators. 1. Nature of Stacking Interactions between Intercalators (Ethidium, Daunomycin, Ellipticine, and 4‘,6-Diaminide- 2-phenylindole) and DNA Base Pairs. Ab Initio Quantum Chemical, Density Functional Theory, and Empirical Potential Study [J]. J Am Chem Soc, 2002, 124: 3366- 3376.
[38] Kurita N, Kobayashi K. Density functional MO calcula- tion for stacked DNA base-pairs with backbones [J]. Comput & Chem, 2000, 24: 351-357.
[39] FukuiK, Yonezawa T, Shingu H. A Molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons [J]. J Chem Phys, 1952, 20: 722-725.
[40] Klopman G. Chemical reactivity and the concept of charge- and frontier-controlled reactions [J]. J Am Chem Soc, 1968, 90: 223-234.
[41] Fleming I. Frontier Orbital and Organic Chemical Reaction [M]. New York: Wiley, 1976.
钌配合物[Ru(phen)2(7-R-dppz)]2+( R = -CH3, -F, -CF3)的合成、表征及DNA键合性质
刘 平1, 刘学文1,2, 陈远道1,2, 卢基林1, 李 琳1
(1. 湖南文理学院 化学化工学院, 湖南 常德, 415000; 2. 湘潭大学 环境友好化学与应用教育部重点实验室, 湖南 湘潭, 411105)
合成并表征了三个钌(II)多吡啶配合物[Ru(phen)2(7-CH3-dppz)]2+(i)、[Ru(phen)2(7-F-dppz)]2+(ii)和 [Ru(phen)2(7-CF3-dppz)]2+(iii). 利用光谱法和粘度实验研究了配合物与DNA的结合行为. 结果表明3个配合物均以插入方式与DNA结合,其与DNA的亲和力顺序为(ii) > (i) > (iii).同时采用密度泛函(DFT)理论计算合理解释了3个配合物与DNA结合行为的差异.
钌(II)配合物;多吡啶配体;DNA结合;密度泛函
2010-06-15
湖南省教育厅科研基金资助项目(10C1002, 04C413); 湖南省自然科学基金资助项目(07JJ3015); 环境友好化学与应用教育部重点实验室开放课题(10HJYH05)
刘平(1956-), 男, 教授, 研究方向为计算化学.
O 614.82+1
A
1672-6146(2010)03-0035-10
10.3969/j.issn.1672-6146.2010.03.012
猜你喜欢
云南化工(2021年10期)2021-12-21
化工学报(2020年4期)2020-05-28
英语文摘(2020年12期)2020-02-06
音乐教育与创作(2019年10期)2019-12-26
今日农业(2019年11期)2019-08-13
活力(2019年21期)2019-04-01
流行色(2017年12期)2017-10-26
发明与创新(2016年7期)2016-12-18
中国塑料(2015年11期)2015-10-14
大众文艺(2015年4期)2015-07-13
