
HPLC-MS/MS测定人体血浆中克拉霉素浓度的方法及方法学确证
2010-03-21 07:28:28张春宝何世学何芳李媛媛陈刚高洪志郝光涛
河北医药 2010年16期
张春宝 何世学 何芳 李媛媛 陈刚 高洪志 郝光涛
克拉霉素(Clarithromycin)是大环内酯类抗生素,适用于革兰氏阳性菌、部分革兰氏阴性菌、厌氧菌及支原体、衣原体等非典型病原体所引起的感染。因其吸收后血药浓度低,有必要建立选择性好、灵敏的分析方法测定其血药浓度,研究其临床药代动力学[1-6]。国内外对生物样本中克拉霉素的测定方法有气相色谱法、液相色谱法、液相色谱-串联质谱联用法等,但很多方法都存在一定的局限性,如样本处理过程复杂,色谱条件复杂(梯度洗脱)等,为了研究克拉霉素磷酸酯片的人体药代动力学,本实验拟建立高效灵敏准确专属性强的方法以测定人血浆中克拉霉素浓度。
1 对象与方法
1.1 仪器与试剂 API3200型三重四极杆串联质谱仪:美国Applied Biosystems公司产品,配有电喷雾离子化源(ESI),色谱工作站为Analyst1.4.2数据处理软件 (美国AB公司),液相色谱仪是岛津(SHIMADZU)高效液相色谱系统,包括二元输液泵、自动进样器、柱温箱。克拉霉素对照品购于中国药品生物制品检定所,含量为99.0%,批号:30356-200403;替米沙坦对照品(内标)由北京昭衍新药研究中心提供,批号:010600160,纯度99.5%。甲醇和甲酸均为色谱纯,为Dikma公司产品;试验用水为去离子水,是本实验室自制;其他试剂为AR试剂。
1.2 溶液的配制
1.2.1 克拉霉素标准溶液的配制:精密称取克拉霉素对照品10.0 mg,置于50 ml容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得200.0μg/ml克拉霉素的标准储备液。
1.2.2 克拉霉素标准溶液的配制:取上述储备液以甲醇依次倍比稀释,配成浓度分别为 10、25、100、200、500、1 500 和3 000 ng/ml,即得。
1.2.3 内标(替米沙坦)标准溶液的配制:精密称取替米沙坦对照品10.0 mg,置于50 m l容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得200.0μg/ml的内标储备液,取该储备液加入甲醇倍比稀释至200 ng/ml,作为内标工作溶液,并将其置于4℃冰箱保存待用。
1.3 血浆样品分析方法 精密量取50μl血浆样品,置于1.5 ml EP试管中,分别加入50μl内标溶液 (200 ng/ml替米沙坦甲醇溶液)、50μl甲醇,并补加350甲醇溶液,涡旋混合30 s,15 000 r/min 低温4℃离心 10 min,取上清液进样 2 μl,进行高效液相色谱-串联质谱联法(HPLC-MS/MS)分析。
1.4 色谱及质谱条件
1.4.1 色谱条件:色谱柱:Agilent-XDB-C8柱 (2.1×150 mm,5 μm),USA; 流动相∶甲醇∶水(含0.1%甲酸)=85∶15(V∶V);流速:0.2 m l/min;柱温:20℃;进样量:2 μl。
1.4.2 质谱条件:离子源:电喷雾离子化源(ESI源);离子极性:正离子(Positive);离子检测方式:多反应监测(MRM)方式;喷射电压:5 500 V;离子源温度:450℃;源内气体1(GS1,N2)压力:40psi;源内气体2(GS2,N2)压力:40psi;气帘气体(N2)压力:15psi;碰撞气(CAD,N2)压力:5 psi;CE电压:分别为50 V(克拉霉素),63 V(替米沙坦,内标);DP电压(解簇电压)分别为55 V(克拉霉素),90 V(替米沙坦,内标);用于定量分析的离子反应对分别为质荷比(m/z)748.4→m/z 158.2(克拉霉素) 和 m/z 515.3→m/z 276.2(替米沙坦,内标)。
2 结果
2.1 质谱分析 取克拉霉素和替米沙坦标准储备液适量用甲醇稀释成浓度为100μg/ml的甲醇溶液,按“1.4.2质谱条件”项操作,相应的二级全扫描质谱图见图1。

图1 克拉霉素和内标替米沙坦的产物离子扫描质谱图
2.2 特异性 分别取6个不同人的空白血浆50μl,除不加内标溶液并另外补加50μl甲醇外,其余按“3血浆样品分析方法”项下方法操作,进样2μl,获得空白样品的色谱图:2-A;将一定浓度的克拉霉素标准溶液和内标替米沙坦溶液加入空白血浆中,依同法进行样品处理,获得相应的色谱图:2-B(最低定量限)、2-C(标准曲线中间浓度)。结果表明,空白血浆中的内源性物质不干扰克拉霉素和替米沙坦的测定。

图2 血浆中克拉霉素和内标替米沙坦的典型MRM色谱图(Ⅰ克拉霉素;Ⅱ替米沙坦)
2.3 相对基质效应考察 分别取6个不同受试者空白血浆(即受试者0 h血浆)50μl于1.5 m l EP试管中,然后各加入克拉霉素最低浓度(10 ng/ml)标准溶液50μl和内标50μl,按“1.3血浆样品分析方法”项下操作,进样量为2μl,每个空白血浆各做3份;并同时做一条随行的标准曲线,根据标准曲线计算血浆样品实测浓度。结果显示所有试验测定值与标示量的相对偏差 (RE%)均在 ±20%之间,精密度为 5.52(<15%),则可认为没有基质效应干扰。
2.4 血浆中克拉霉素标准曲线制备及定量下限的确定 取人空白血浆50μl,分别精密加入标准系列溶液的克拉霉素对照品溶液50μl和内标溶液50μl,旋涡混匀,配成含克拉霉素的浓度分别为 10、25、100、200、500、1 500 和 3 000 ng/ml的标准含药血浆,按“3血浆样品分析方法”项下操作,记录色谱图,计算克拉霉素峰面积As和内标峰面积Ai的比值f(f=As/Ai)。以血浆中待测物浓度(X)为横坐标,待测物与内标的峰面积比值(Y)为纵坐标,用加权最小二乘法进行回归运算,求得回归方程(图3):Y=0.00257X+0.00441,r=0.9989,权重系数w=1/χ2。按上述条件克拉霉素在血浆中的定量下限(LLOQ)为10 ng/ml。配制克拉霉素浓度为10 ng/ml血浆样品5份,进行HPLC-MS/MS分析,记录色谱图,计算克拉霉素峰面积As和内标峰面积Ai的比值f,按照当日随行标准曲线方程求得实测浓度及实测浓度准确度,并计算RSD,结果表明所有试验测定值与标示量的相对标准偏差(RSD%)均在±15%之间。
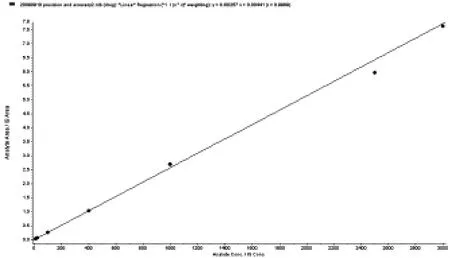
图3 HPLC-MS/MS法测定人血浆中克拉霉素的标准曲线
2.5 精密度试验 取1.5 ml EP试管数支,按“2.4血浆中克拉霉素标准曲线制备及定量下线的确定”项制备含克拉霉素浓度分别为25、400、2 500 ng/ml的标准含药血浆(每个浓度做6份样品)及一条随行标准曲线,按“1.3血浆样品分析方法”项下操作。每天做一批及一条随行标准曲线,连续做3 d,共三批,每批每个浓度做6份样品,记录色谱图,计算克拉霉素峰面积As和内标峰面积Ai的比值f,代入当天的标准曲线求得实测浓度,计算批内和批间精密度。见表1。

表1 HPLC-MS/MS法测定血浆中克拉霉素的批内和批间精密度n=18
2.6 准确度试验(相对回收率) 取1.5 m l EP试管数支,按“2.4血浆中克拉霉素标准曲线制备”项制备含克拉霉素浓度分别为25、400、2500 ng/ml的含药血浆,按“3血浆样品分析方法”项下操作,记录色谱图,计算克拉霉素峰面积As和内标峰面积Ai的比值f,将比值f代入当天的标准曲线方程,计算实测浓度与加入浓度的比值即为准确度(相对回收率)。血浆中低、中、高3个浓度克拉霉素的提取回收率分别为(94.1±8.00)%、(99.6 ±8.20)%、(97.3 ±4.70)%,平均提取回收率为(97.0 ±6.97)%。
2.7 样品稳定性考察 分别考察:克拉霉素血样处理后的检测样品进样器中放置4 h稳定性;血浆样品室温放置4 h稳定性;血浆样品-40℃反复冻融3次稳定性;血浆样品-40℃冻存1个月后稳定性。即制备低、中、高(25、400、2 500 ng/m l)3个浓度的质控样品,每个方法的每个浓度个3份,按“1.3血浆样品分析方法”项下操作,每个方法最后所得上清液作为对应0 h的检测样品溶液,立即取5μl进样分析,按照当日标准曲线求出各样品的浓度,计算各样品的测得值与实际值的相对偏差(RE%),与0 h的相应浓度的样品比较。见表2。
3 讨论
HPLC-MS/MS法具有选择性好、灵敏度高、高通量等优点,目前已被广泛应用于检测生物样本中的药物以及药代动力学等研究,而基质效应则是此法测定生物样本中药物浓度时十分重要的问题,完整的方法确证应包括对于方法基质效应的研究。采用梯度洗脱的色谱分离方法分析测定时间过于长,影响了方法的测定效率[6-8]。Cherlet指出使用高比例有机相进行洗脱[7],虽然克拉霉素能较快洗脱出峰,但同时也受到快速洗脱出的基质对其离子化的影响。
本研究建立了测定人血浆中克拉霉素的HPLC-MS/MS法,并进行了系统的方法确证,沉淀蛋白前处理方法简单快速,优化后的色谱条件极大缩短检测时间,同时也减少了沉淀蛋白后生物样本中基质对克拉霉素的影响,实现了高通量测定,本方法灵敏度高,血浆用量仅为50μl,定量下限为10 ng/ml,能够满足克拉霉素的人体药代动力学研究的需要。
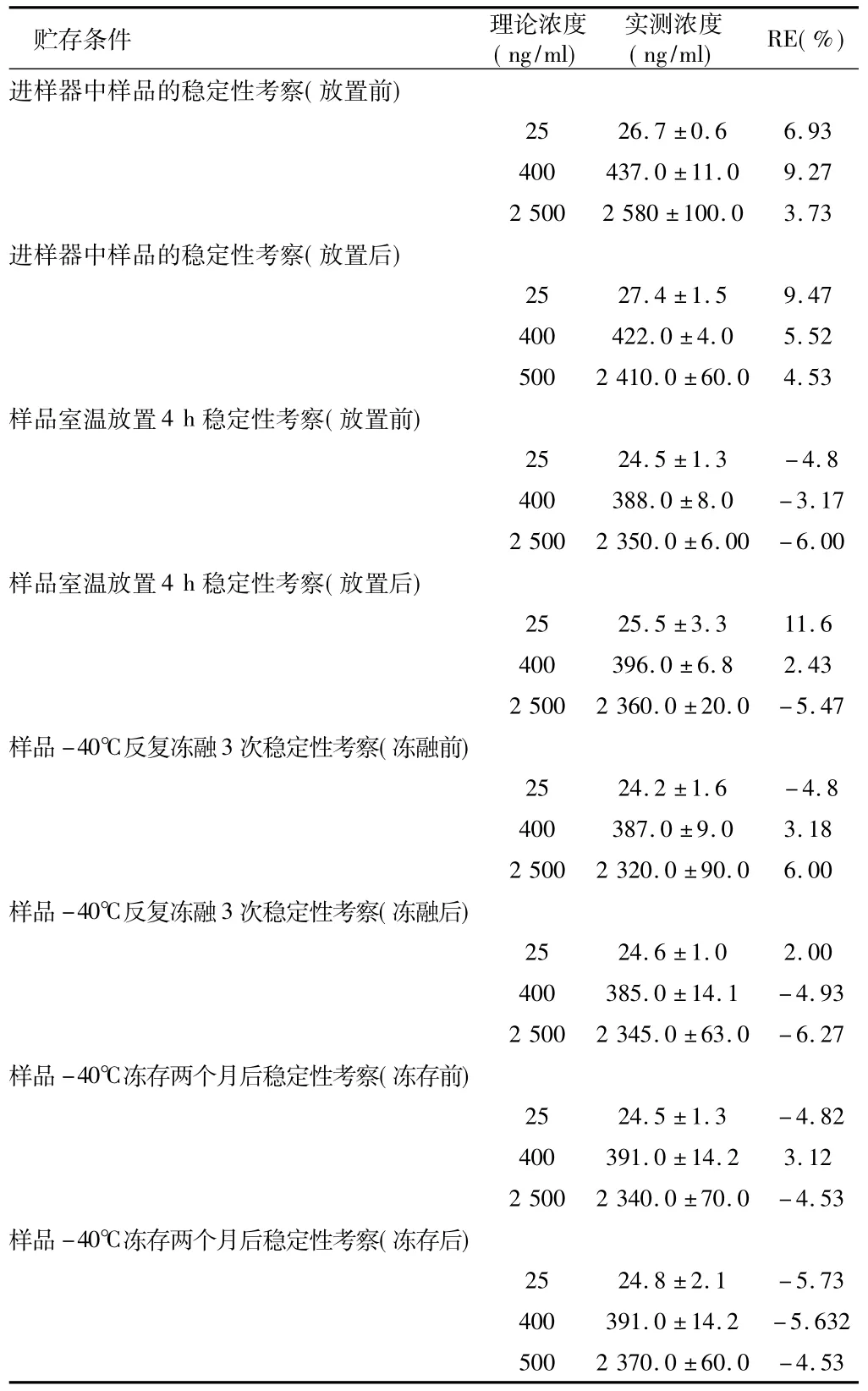
表2 各种贮存条件下血浆中克拉霉素稳定性试验 n=3
1 国家药典委员会.中华人民共和国药典(2000年版二部).北京:化学工业出版社,2000.附录193-197.
2 李军,张晓坚,刘广炼.克拉霉素药代动力学的研究.数理医药学杂志,2000,13:244-245.
3 俞永进.HPLC内标法测定克拉霉素含量.中国药房,1999,10:137-138.
4 王庆全,张玉国.HPLC法测定克拉霉素颗粒剂的含量.中国抗生素杂志,2002,27:628-629.
5 莫宗琪,吴金虎,裴琳,等.克拉霉素的高效液相色谱法与微生物效价测定法分析比较.中国医院药学杂志,2002,22:378-379.
6 Cherlet M,Croubels S,De Backep P.Determination of clindamycin in animal plasma by high-performance liquid chromatography combined with electrospray ionization mass spectrometry.J Mass Spectrom,2002,37:848-853.
7 Yu LL,Chao CK,LiaoWJ,et al.Determination of clindamycin in animal plasma by chromatography-electrospray tandem mass spectrometry:application to the bioequivalence study of clindamycin.JChromatogr B,1999,724:287-294.
8 Li J,Wang N,Zhang ZJ,etal.Pharmacokineticsand bioequivalence study of clindamycin hydrochloride formulations after single-dose administration in healthy Chinese male volunteers.Arzneimitte Forschung,2008,58:358-362.
猜你喜欢
中国宝玉石(2021年5期)2021-11-18 07:42:26
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
世界最新医学信息文摘(2021年12期)2021-06-09 08:37:02
中国特种设备安全(2021年12期)2021-04-26 14:37:00
大众电视(蓝天下)(2018年8期)2018-10-26 01:00:48
中成药(2018年6期)2018-07-11 03:01:32
超硬材料工程(2016年1期)2016-02-28 22:20:07
中国粮油学报(2016年5期)2016-01-23 02:45:06
中国当代医药(2015年16期)2015-03-01 02:03:19
中国当代医药(2015年7期)2015-03-01 02:01:22
