
n-C3H7I和i-C3H7I在266 nm的光解:烷基自由基分支化对C—I解离动力学的影响
2010-03-06 04:44王艳梅冯文林
物理化学学报 2010年7期
张 锋 王艳梅 张 冰,* 冯文林
(1重庆理工大学光电信息学院,重庆 400051; 2中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉 430071;3中国科学院研究生院,北京 100049)
Alkyl halides are particularly attractive in the study of photodissociation reactions since they provide the opportunity to understand the effect of size and symmetry of the alkyl radicals on the dynamics of bond fission.Photodissociation dynamics of simple alkyl halides in the A-band arising from σ*←n transition of a lone pair electron of halogen atoms has attracted considerable attention in the past several decades[1-7].Generally,the excitation to the A-band of a molecule causes the fission of C—X(X is halogen atom)bond to produce an alkyl radical with the responding halogen atom X(2P3/2)or X*(2P1/2).Three states,3Q1,3Q0and1Q1in Mulliken′s notation,in the σ*configuration are dipole allowed from the ground state of alkyl halides and are repulsive in nature.The3Q0state correlates with the X*fragment while the other two states lead to the X formation.The final yields of X and X*from the A-band photodissociation of the molecule are mediated by the nonadiabatic transition between the3Q0and1Q1states.
As the simplest alkyl halide,photodissociation of methyl halides in the A-band has been a paradigm for experimental[8-10]and theoretical researches[11-15]on photodissociation processes that occur along a repulsive potential surface of the excited state, by which the molecule directly dissociates into CH3and halogen atom[8-12].For instance,dissociation dynamics of methyl iodide in C3vgeometry can be relatively well confined within the ν2umbrella and ν3symmetric stretch modes,under the simplified assumption of collinear pseudotriatomic dissociation due to prompt dissociation[13].In fact,the computation of the three dimensional photodissociation dynamics of the selected rotational state shows that the overall rotation has significant effects on the methyl rotational and vibrational distributions as well as the I*yield[14].An ab initio study indicates that vibrational state control of the I*/I branching ratio in the alkyl(hydrogen)iodide photodissociation has an electronic rather than a dynamic nature[15].The study of a series of alkyl iodides[16-17]using resonance Raman spectra shows that bend-stretch combination band progressions exist in addition to C—I stretch normal modes.The intensity of bend-stretch combination band progressions increases with the alkyl radicals being heavier and more branched relative to the C—I stretch normal modes.This suggests that the photodissociation along reaction coordinate has significant multidimensional character coded with more complicated vibration and rotation modes.
The photodissociation of n-C3H7I and i-C3H7I offers a good choice to check the effects of the geometry of an alkyl radical on photodissociation since these two molecules are isomers with the different radical branching at α-carbon atom.Taking advantage of ion imaging technique in this work,we investigated the photolysis of n-C3H7I and i-C3H7I at 266 nm.With the information extracted from the energy and angular distributions of photoproducts I and I*of these two molecules,we gave quantitatively the cross sections and the crossing probability between different dissociative states.Finally,these results are compared to gain insight into the effect of radical branching on C—I dissociation dynamics.
1 Experimental
The experiments were performed on a home-built velocity ion imaging apparatus that was described in detail elsewhere[18]. Briefly,the apparatus consists of three parts:a source chamber, a main chamber,and a detector.Both of the chambers were pumped to obtain a pressure of about 5.0×10-6Pa.A molecular beam was produced through a pulsed valve synchronized with the laser pulses at 10 Hz and intersected with linearly polarized tunable ultraviolet(UV)laser pulses in main chamber through a conical skimmer.The generated ions were extracted and accelerated by the electrostatic immersion lens and projected onto a two-dimensional(2D)detector consisting of two microchannel plates coupled with a P47 phosphor screen and a charge-coupled device camera.
The UV laser pulses were frequency doubled output of a dye laser system pumped by the harmonic of a Nd:YAG(YG980, Quantel,France)operating at 10 Hz and were used to dissociate molecules.Within the same pulse,the atomic fragments were state-selectively ionized by a(2+1)REMPI process via 7p(4S3/2)←5p5(2P3/2)transition for I(2P3/2)and np(73/2)←5p5(2P1/2)transition for I*(2P1/2)[19].Each ion image was constructed by accumulating signals from 10000 shots of the pulse and scanning the laser wavelength over the entire Doppler profile of the detected species.In order to minimize the influence of clusters,photolysis was performed on the rising edge of the molecular beam pulse.During the experiments,all the time delays were controlled by a pulse delay generator(Stanford DG535 Pulse Generator,SRS Inc., USA).
The liquid samples of n-C3H7I and i-C3H7I with the purity of 99.9%were seeded in Helium gas at 1.0×105Pa without further purification and then were introduced into the source chamber through the pulsed valve.
2 Results
Fig.1 shows the images of I(2P3/2)and I*(2P1/2)atoms from the photodissociation of n-C3H7I at 266 nm with the laser beam polarized along the vertical direction.Background counts in the raw images have been removed by subtracting the reference image acquired at off-resonance wavelength of iodide atoms under the same conditions.The distribution of fragments in space is cylindrically symmetric about the polarization axis of photolysis laser and can be reconstructed from the raw images by the basisset expansion method(BASEX)[20].The reconstructed images are also showed in Fig.1.Similarly,Raw images and the corresponding reconstructed images of I and I*from the photodissociation of i-C3H7I at 266 nm are displayed in Fig.2.
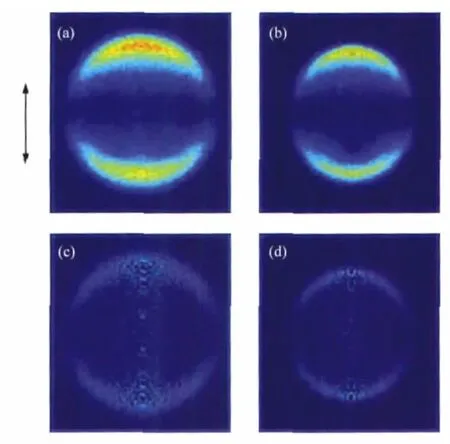
Fig.1 Raw images of I(a)and I*(b)and the corresponding reconstructed ones of I(c)and I*(d)fragments from the photodissociation of n-C3H7I at 266 nmThe arrow denotes the polarization vectors of the photolysis laser.
From the reconstructed ion images,the speed distributions, P(v),can be derived by integrating over all angles at each speed. Thus,the total translational energy(ET)distribution of each photodissociationchannelinmolecularcoordinate,P(ET),asshownin Fig.3,can be easily obtained from P(v)of an individual fragment by the following formulas:
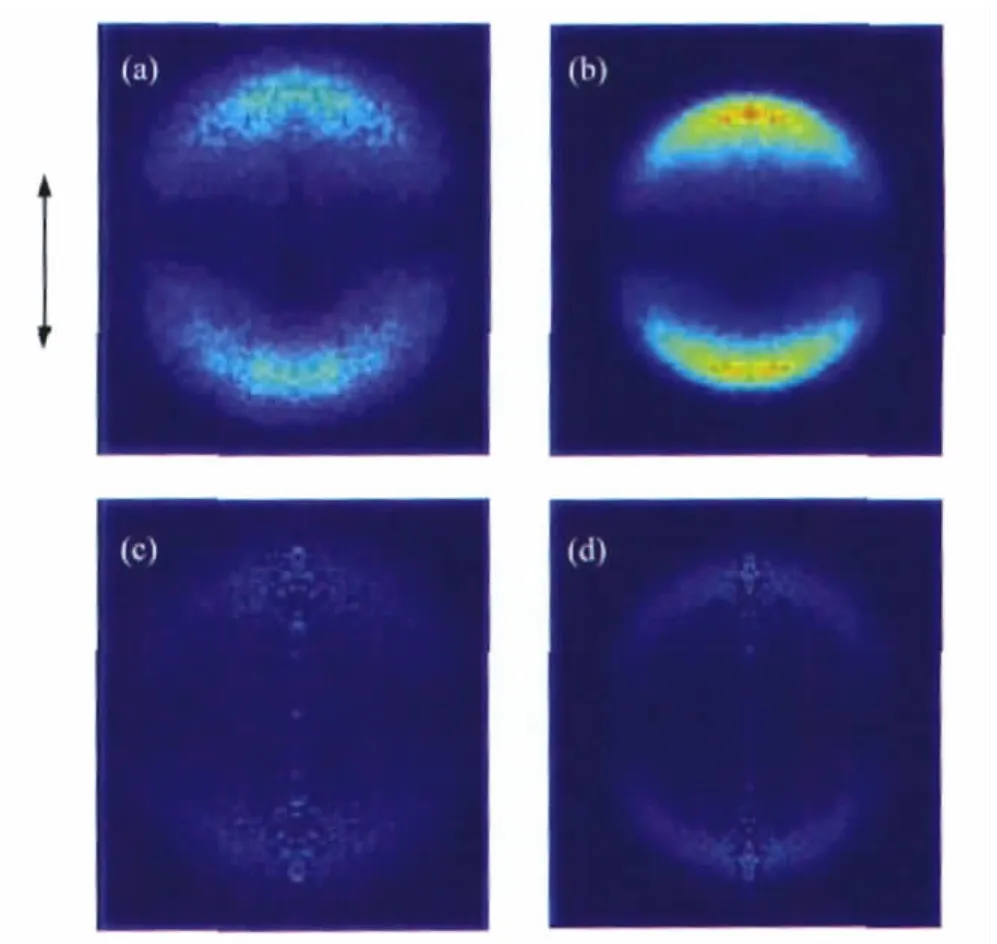
Fig.2 Raw images of I(a)and I*(b)and the corresponding reconstructed ones of I(c)and I*(d)fragments from the photodissociation of i-C3H7I at 266 nmThe arrow denotes the polarization vectors of the photolysis laser.
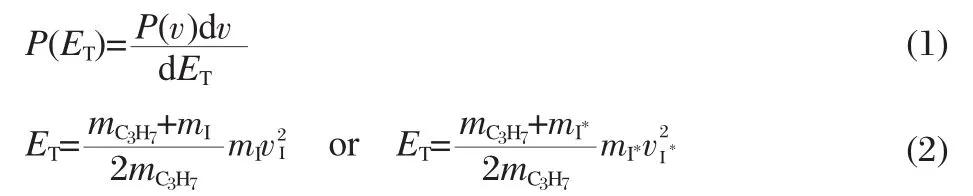
where dv and dETare the differential elements for the velocity v and the total translational energy ET,respecitively,mxand vx(x= C3H7,I and I*)are the mass and velocity of the photoproducts, respectively.The average translational energy〈ET〉and the corresponding full width at half maximum(FWHM)of I and I*cha-nnels of these two molecules are abstracted from the Gaussian fitting functions and are listed in Table1.According to energy conversation,the maximum available energy for the dissociation process Eavlis calculated by the following equation:

Fig.3 Total translational energy distributions(P(ET))of the I and I*channels of the n-C3H7I(a,b)and i-C3H7I(c,d)at 266 nmThe circles represent the experimental results and the solid lines are the best-fitting of the experimental data.

where Einis the initial internal energy of the parent molecules, Ehνis the pump photon energy,D0is the C—I bond dissociation energy of parent molecules at the ground states,228 kJ·mol-1for n-C3H7I and 221 kJ·mol-1for i-C3H7I[2],Eelis the electronic energy of iodine atom,zero for I and 91 kJ·mol-1for I*.Einis very small for the supersonic molecular beam and assumed to be zero.The fraction of the translational energy,defined as fT=〈ET〉/ Eavlfor I or I*dissociation channel,is also determined.These energy values are listed in Table1.
Angular distributions of the fragments I and I*,I(θ),are extracted by integrating the reconstructed three-dimensional spatial distribution over a proper range of speed at each angle.Generally,it may be characterized by anisotropy parameter β as expressed by Eq.(4):

where P2(cosθ)is the second order Legendre polynomial,and θ is the angle between the recoil velocity vector of fragments and the pump polarization axis.The measured β values here are averaged over the range of the FWHM of the translational velocity distributions and are listed in Table 2.For a particular excited state in which a molecule dissociates quickly along a bond axis, the anisotropy parameter β can also be given by Eq.(5):

where P2(cosχ)is the second order Legendre polynomial and χ is the angle between the transition dipole moment of an excited state and bond axis.It can be seen that β varies from+2(the limit of a parallel transition(χ=0°))to-1 for that of a perpendicular transition(χ=90°).The envelopes of the ultraviolet absorption spectrum of both n-C3H7I and i-C3H7I[21]are similar to that of CH3I and show a Gaussian-type broadband centered at ca 255 nm and ca 260 nm,respectively.These absorption broadbands can be resolved into the3Q1,3Q0and1Q1states analogous to that of CH3I in energy ascending order.Among these states,3Q0transition dominates the absorption band[1-2,14-15].In our experiment, the optical absorption at 266 nm,being in the red wing of the absorption spectra,should be accompanied with the transitions to the lower lying3Q1and3Q0states.The3Q0state correlates not only to the I*fragments but also to the I fragments through a nonadiabatic process due to curve crossing between the3Q0and1Q1states.The3Q1state directly correlates to I fragments.Thus, anisotropy distributions of I*fragments along the polarization of pump laser reflect the angle χ between the3Q0transition dipole moment and C—I bond axis of n-C3H7I or i-C3H7I,which is evaluated to be 15°for n-C3H7I and 18°for i-C3H7I from their individual β(I*)values according to the Eq.(5).

Table 1 Values of the energy parameters for I and I* channels from the photodissociation of the n-C3H7I and i-C3H7I at 266 nm

Table 2 Anisotropy anisotropy parameter,relative oscillator strengths,and fraction of a wavepacket along the possible dissociation potential energy surface at 266 nm for n-C3H7I and i-C3H7I,C2H5I,and CH3I
3 Discussion
The images of I and I*of both n-C3H7I and i-C3H7I show simple structures(Fig.1 and Fig.2)with sharp anisotropydistributions along the polarization of pump laser,suggesting that the C—I bond dissociation promptly happens within a rotation period. The distributions can be well fitted by a single-peaked Gaussian curve as shown in Fig.3.We obviously can see that the energy distributions of I fragments of both molecules are much wider than those of I*fragments,which reflects that the radicals accompanied with I fragments should be in hotter internal states than those produced by dissociation channel n-or i-C3H7+I*. This is a common character of the photodissociation dyanmics of all simple alkyl halides because of the greater available energy for the radical+X(2P3/2)reaction[1-2,18,22],in which more rotation and vibration modes of the radicals are excited.This is also confirmed by the fact that the translational energy fraction(fT)of I*fragments is larger than that of I fragments for both molecules as listed in Table 1.
The resonance Raman spectra of a series of alkyl iodides[16-17]at 266 nm have shown that as the alkyl radical becomes heavier and more branched,the Raman spectra show increased intensity in bend-stretch combination band progressions relative to the C—I stretch overtone progression.Namely,the dissociation coordinate is not only along the C—I internal coordinate but also along the bending internal coordinates with increase of the mass and branches of the alkyl radical.Comparing n-C3H7I with i-C3H7I,from Fig.3 and Table 1 we can also see that the energy distributions of the same iodine atom become wider and the corresponding translational energy fractions become smaller with the radical on α-carbon atom being more branched.In the impulsive framework,the width of the energy distribution is a reflection of the initial spread of momenta of the C—I caused by local excitation to the C—I bond.Those atoms attached to the α-carbon have a significant effect on the initial spread of momenta of the C—I bond.That means that more branched and heavier are the radicals attached to the α-carbon,more easily happen bend vibration of the C—C—C chains for the n-C3H7I and i-C3H7I.
As described above,the anisotropydistribution parameters β(I*) of the I*products from these two isomers directly reflect the alignment of the transition dipole of the3Q0state along the C—I bond axis.However,the I product originates from two channels: direct excitation of the3Q1state and the1Q1←3Q0nonadiabatic transition.The direct contribution in the I channel is considered to be the reflection of the alignment of the transition dipole of the3Q1state to the C—I bond axis,while the nonadiabatic contribution remains the same anisotropy as the fragments that appear in the I*channel.Therefore,the β(I)value may be resolved to the relative contributions of the3Q1and3Q0states by Eq.(6)[23]:

where P3Q0or P3Q1represents a relative oscillator strength for the transition to the3Q0and3Q1states,respectively,andf(3Q0,I)and f(3Q0,I*)(in Eq.(7))are the fractions ofthe wavepackets going into the I and I*channels after pumped to the3Q0state, respectively,and f(3Q0,I)+f(3Q0,I*)=1.β3Q0and β3Q1represent the effective anisotropy parameter limit for the alignment of3Q0and3Q1transitions,respectively.and-0.90 for n-C3H7I and β3Q0=β(I*)=1.71 andfor i-C3H7I.In addition,the P and f are connected by Eq.(7)[23]:

where Φ(I*)/Φ(I)is the ratio of relative quantum yields of the I*and I from the dissociation of a molecule,and have been exactly determined to be 0.7/0.3 and 0.44/0.56 for n-C3H7I and i-C3H7I[24],respectively.All the values calculated here are listed in Table 2.Obviously,the relative oscillator strengths of the transition to3Q0states are absolutely dominated,0.97 for n-C3H7I and 0.95 for i-C3H7I.Using magnetic circular dichroism technology, Gedanken et al.[25-26]also revealed that the3Q0←X transition carries over 80%oscillator strengths in the A-band spectra of these alkyl iodides[25-26].The difference of these two molecules is small in oscillator strengths of the transition from the ground state to3Q0state.However,the wavepackets on the3Q0state prepared by a 266 nm photon proceed nonadiabatically to give the I*fragment with a probability f(3Q0,I*)of 0.72 for n-C3H7I and 0.46 for i-C3H7I,the remaining wavepackets product the I fragment with the probability of 0.28 for n-C3H7I and 0.54 for i-C3H7I,respectively.
The effects of radical size and branching on upper state symmetry and curve crossing probabilities are obvious by comparing our data with those for CH3I[9]and C2H5I[24]as listed in Table 2. Though the relative oscillatorstrengthsofthese moleculespumped by 266 nm photon display a small difference for the3Q0←X transition,the probability to yield I fragment via curve crossing between the3Q0and1Q1state shows a large difference as the radicals being more branched:0.54 for i-C3H7I and 0.26-0.29 for other n-alkyl iodides.This fact reflects that the introduction of the methyl to α-carbon atom enhances the coupling strengths between3Q0and1Q1states,which makes the probability of yielding I fragments increase after a wavepacket is pumped to3Q0state by 266 nm photon.Photodissociation of these alkyl iodides at 248 nm have been investigated using translational photofragment spectroscopy by Godwin and his co-workers[1-2].The fraction of the I quantum yield from i-C3H7I is obviously larger than that from other n-alkyl iodides.The results reported by Phillips et al.[16-17]have shown that the unique structure of the radical from i-C3H7I exhibits the stronger bend-stretch combination band relative to n-C3H7I.Moreover,as the alkyl radical becomes heavier and more branched the bend-stretch combination band progres-sion of the molecules becomes more obvious relative to the C—I stretch overtone progression.The formation of this enhanced bend-stretch combination band can cause the increase of the coupling strength between3Q0and1Q1states and should be responsible for the enhancement in the crossing probability that a wavepacket from3Q0goes into the I channel.
4 Conclusions
The photodissociation dynamics of n-C3H7I and i-C3H7I at 266 nm was investigated using ion imaging detection.For both molecules,the dissociation products are iodide atom(I or I*) and the responding alkyl radical with a single energy distribution.The energy disposed into internal motions of the molecules for I channel is greater than that for I*channel because of the greater available energy,with which complex vibration and rotation modes can be excited more easily.As the alkyl group becomes more branched,the mixing of the rovibrational motions about the α-carbon atom with the C—I stretching in the photodissociation of alkyl iodides becomes more significant.Though the relative oscillator strengths of these molecules pumped by 266 nm photon display a small difference for the3Q0←X transition,the probability to yield I fragment via curve crossing between the3Q0and1Q1shows a large difference:0.54 for i-C3H7I and 0.26-0.29 for other n-alkyl iodides.It is proposed that the contribution of bending motion of the molecule becomes more significant and the coupling strength between3Q0and1Q1states gets stronger greatly during the C—I dissociation.In addition, the3Q0←X transition is not completely parallel transition for both molecules and the angle between the transition dipole moment and bond axis is estimated to be about 15°for n-C3H7I and 18°for i-C3H7I,respectively.
1 Paterson,C.;Godwin,F.G.;Gorry,P.A.Mol.Phys.,1987,60: 729
2 Godwin,F.G.;Paterson,C.;Gorry,P.A.Mol.Phys.,1987,61: 827
3 Zhu,Q.H.;Cao,J.R.;Wen,Y.;Zhang,J.M.;Zhong,X.;Huang, Y.H.;Fang,W.Q.;Wu,X.J.Chem.Phys.Lett.,1988,144:486
4 Matsumi,Y.;Tonokura,K.;Kawasaki,M.J.Chem.Phys.,1991, 94:2669
5 Uma,S.;Das,P.K.J.Chem.Phys.,1996,104:4470
6 Underwood,J.G.;Powis,I.Phys.Chem.Chem.Phys.,2000,2: 747
7 Zhou,J.G.;Lau,K.C.;Hassanein,E.;Xu,H.F.;Tian,S.X.; Jones,B.;Ng,C.Y.J.Chem.Phys.,2006,124:034309
8 Hertz,R.A.;Syage,J.A.J.Chem.Phys.,1994,100:9265
9 Eppink,A.T.J.B.;Parker,D.H.J.Chem.Phys.,1998,109:4758
10 Samartzis,P.C.;Bakker,B.L.G.;Parker,D.H.;Kitsopoulos,T. N.J.Phys.Chem.A,1999,103:6106
11 Rist,C.;Alexander,M.H.J.Chem.Phys.,1993,98:6196
12 Thanopulos,I.;Shapiro,M.J.Chem.Phys.,2006,125:133314
13 Alekseyev,A.B.;Liebermann,H.P.;Buenker,R.J.;Yurchenko,S. N.J.Chem.Phys.,2007,126:234102
14 Xie,D.Q.;Guo,H.;Amatatsu,Y.;Kosloff,R.J.Phys.Chem.A, 2000,104:1009
15 Alekseyev,A.B.;Liebermann,H.P.;Buenker,R.J.J.Chem. Phys.,2007,126:234103
16 Phillips,D.L.;Lawrence,B.A.;Valentini,J.J.J.Phys.Chem., 1991,95:7570
17 Phillips,D.L.;Lawrence,B.A.;Valentini,J.J.J.Phys.Chem., 1991,95:9085
18 Tang,Y.;Ji,L.;Tang,B.F.;Zhu,R.S.;Zhang,S.;Zhang,B.Acta Phys.-Chim.Sin.,2004,20(4):344 [唐 颖,姬 磊,唐碧峰,朱荣淑,张 嵩,张 冰.物理化学学报,2004,20(4):344]
19 Donovan,R.J.;Flood,R.V.;Lawley,K.P.;Yencha,A.J.;Ridley, T.Chem.Phys.,1992,164:439
20 Dribinski,V.;Ossadtchi,A.;Mandelshtam,V.A.;Reisler,H.Rev. Sci.Instrum.,2002,73:2634
21 Roehl,C.M.;Burkholder,J.B.;Moortgat,G.K.;Ravishankara,A. R.;Grutzen,P.J.J.Geophys.Res.,1997,102:12819
22 Wang,Y.;Zhang,S.;Wei,Z.;Zheng,Q.;Zhang,B.J.Chem. Phys.,2006,125:184307
23 Lee,K.S.;Lim,J.S.;Ahn,D.S.;Choi,K.W.;Kima,S.K. J.Chem.Phys.,2006,124:124307
24 Fan,H.Y.;Pratt,S.T.J.Chem.Phys.,2005,123:204301
25 Gedanken,A.;Rowe,M.D.Chem.Phys.Lett.,1975,34:39
26 Gedanken,A.Chem.Phys.Lett.,1987,137:462
猜你喜欢
中国机械工程(2022年18期)2022-10-08
波谱学杂志(2021年3期)2021-09-07
初中生世界(2021年14期)2021-04-15
初中生世界(2021年10期)2021-03-15
初中生世界(2021年9期)2021-03-15
初中生世界(2020年34期)2020-09-16
中国机械工程(2019年19期)2019-10-28
中国机械工程(2019年11期)2019-06-13
中国机械工程(2019年4期)2019-03-06
浙江工业大学学报(2017年5期)2018-01-22
