
微阵列比较基因组杂交技术检测不明原因智力低下/发育迟缓患儿的基因组拷贝数变异
2010-01-23 03:37陈晓丽王立文丁秀原吴柏林
中国循证儿科杂志 2010年2期
陈晓丽 郭 金 王 珺 王立文 丁秀原 张 霆 吴柏林
儿童发育迟缓(developmental delay,DD)和智力低下(mental retardation,MR)发病率约为3%[1]。尽管传统细胞遗传学检测(常规染色体G带分析)、FISH和新近发展的多重连接依赖探针扩增法(multiplex ligation-dependent probe amplification, MLPA)等技术可提高MR/DD患儿的病因检出率,但仍有50%的患儿病因不明[2]。近年来,随着微阵列技术分辨率的提高,国外科研人员利用微阵列比较基因组杂交(array-comparative genomic hybridization, Array-CGH)技术对不明原因MR/DD患儿开展了全基因组拷贝数变异(copy number variations, CNVs)的研究,发现部分MR/DD患儿存在罕见CNVs,从而识别出一系列新的微缺失或重复综合征[3,4]。中国至今尚未见这方面的研究报道。中国儿童MR/DD患病基数大,是神经科主要就诊人群,其中至少有2/3病因不明[5]。本研究应用高分辨Array-CGH技术,对不明原因MR/DD患儿进行了初步的全基因组CNVs筛查,了解可能与MR/DD相关的罕见基因组CNVs在中国人群不明原因MR/DD患儿中的检出率,以此评估Array-CGH对不明原因MR/DD可能的遗传病因诊断作用。
1 方法
1.1 研究对象
1.1.1 MR的诊断标准 根据不同的年龄段,分别采用《儿心0~4岁精神发育量表》和《中国韦氏智力量表》测定儿童的发育商(DQ)或智商( IQ ),同时运用《婴儿-初中生社会适应性能力量表》进行社会适应性能力评价。IQ或DQ<70,同时伴社会适应性能力低下者诊断为MR;>5岁称为MR,<5岁称为DD;以下统称为MR/DD。
1.1.2 研究对象筛选标准 不明原因的MR/DD患儿指排除出生时产伤、生后中枢神经系统感染和(或)头颅损伤、染色体G带检查异常和(或)其他已知遗传性综合征和遗传病,同时符合以下至少1个条件:①伴发有其他体表和(或)内脏畸形,如先天性心脏病、脊柱畸形、多或少指(趾)等;②母亲具有异常妊娠史,包括多次流产、胎死宫内、死产或出生缺陷;③患儿诊断为孤独症或具有孤独症谱系障碍(autism spectrum disorder, ASD);④患儿具有典型面部特征或异常面容;⑤患儿存在严重的生长落后或发育迟缓。
1.1.3 研究对象剔除标准 所有患儿完成简单尿液筛查以排除遗传代谢性疾病,针对某些疑似综合征患儿,如脆性X染色体综合征(fragile X syndrome)或 Rett综合征, 采用Southern Blotting或PCR、MLPA进行排除检查。
1.1.4 伦理审核 所有患儿均为临床病例,患儿监护人填写书面知情同意书或口头同意参加全基因组CNVs检测。本研究获得首都儿科研究所伦理委员会批准。
1.2 Array-CGH检测方法 按照美国波士顿儿童医院临床分子诊断实验室的标准方法进行操作[6]。QIAGEN 抽提外周抗凝血的基因组DNA,获得吸光度值和浓度后,取3~6 μg DNA于50 μL体系,37℃消化2 h(Alu Ⅰ和Rsa Ⅰ联合消化); QIAprep Spin Miniprep Kit进行DNA纯化,重新测定吸光度值后,取2 500 ng DNA, 用BioPrime labeling kit进行荧光标记(cy5标记对照标本,cy3标记MR/DD标本),37℃ 2 h后终止反应;再次MicroCon YM-30 纯化荧光标记DNA后,将对照DNA和患儿DNA等量混合后加入杂交体系,94℃变性,37℃ 0.5 h,上样到Oligo 244 K芯片上,65℃杂交炉孵育72 h后先用洗脱液1洗脱5 min,再用洗脱液2洗脱1 min, 迅速进行微阵列扫描,采用Feature Extraction 9.0进行数据提取至DNA analystic 5.0软件进行CNVs分析。以上仪器和试剂均有商品化供应(Invitrogen和Agilent公司)。男女性对照DNA样本购自Invitrogen公司。
针对所发现的罕见CNVs,尽可能采用双亲样本重复Array-CGH检测,了解其是否为新生(De novo)CNV。
1.3 MR/DD相关CNVs 的评估方法 针对所有发现的CNVs,首先将这些CNVs与国际基因组CNVs多态性数据库(database of genomic variants,DGV)和UCSC brower(build 18)比对,了解其是否为罕见CNVs(rare CNVs)或常见CNVs(common CNVs),然后根据美国波士顿儿童医院遗传诊断实验室的CNVs 评估分析标准将CNVs分为6类(图1):①常见CNVs:至少有2例以上片段大小几乎完全相同的CNVs或者80%以上区域完全重叠的相似CNVs,曾在DGV和UCSC brower中有报道;②罕见CNVs,且目前已明确与MR/DD相关(已发现的微缺失/重复综合征);③罕见CNVs,但很可能与MR/DD相关, 曾被DECIPHER数据库、美国波士顿儿童医院遗传诊断实验室的aCGH-CNVs 数据库报道,或被既往的MR/DD微阵列基因组研究文献所报道,且已报道的CNVs大小几乎完全相同或者至少80%区域重叠;④新生CNVs:在正常父母样本中未曾发现,该 CNVs虽然在研究文献中未曾报道,但含有与MR/DD可能相关的重要基因;⑤临床意义不明确的CNVs:该CNVs未曾报道,且父母未能参与研究;⑥家族性CNVs:已在正常父母样本中证实。 以上分类②~④均属于与MR/DD相关或很有可能相关的CNVs,对于此类CNVs,该区域内的基因名称来自UCSC brower(build 18)。

图1 CNVs 评估分析平台
Fig 1 The workflow for CNVs evaluation
Notes MR:mental retardation;DD:developmental delay
1.4 文献比对分析 在PubMed数据中,采用“unknown” or “unexplained”、“mental retardation” or “developmental delay”和“array”作为检索词,并组合成不同的检索式进行检索。时间为2006年1月1日至2009年7月31日,检索到一系列利用微阵列技术对不明原因MR/DD人群进行全基因组CNVs研究的文献,比对分析以确认本研究所发现的CNVs 在既往研究中是否有报道。涵盖的研究方法不局限于Oligo CGH array,还包括BAC-CGH array和SNP array。
2 结果
2.1 研究对象的一般情况 2004年7月至2008年7月在首都儿科研究所共收集不明原因MR/DD患儿111例,均为汉族,平均年龄为6岁(1个月至16岁),男女比例为1.775(71∶40), 86例(77.4%)伴有生长落后或发育迟缓;29例(26.1%)伴有其他畸形和(或)神经系统异常,包括先天性心脏病,多指或趾,骨骼或脊柱异常,惊厥,刻板或孤独样症状;13例(11.7%)患儿的母亲具有异常妊娠史;32例(28.8%)患儿具有MR/DD家族史;45例(40.5%)患儿有典型面部特征。37/111例患儿完成了常规染色体G带检查,均提示正常染色体核型。
2.2 Array-CGH的结果及数据比对分析 28/111例患儿中发现36个CNVs(重复16个,缺失20个),发生概率为32.4%(36个/111例),CNVs平均长度为1 326 kb(29~8 760 kb)(表1)。
4/28例患儿获得父母血样进行亲本鉴定,以了解患儿的CNVs是否为家族性遗传。发现2例患儿具有新生CNVs;另2例为父亲遗传,其中1例遗传自智力或神经、精神发育缺陷的父亲,另1例父亲的智力或神经、精神发育情况不详。
对28例患儿所发现的36个罕见CNVs,利用美国波士顿儿童医院遗传诊断实验室的CNVs评估分析平台,结合DECIPHER数据库和既往MR/DD相关的Oligo CGH array、BAC-CGH array和SNP array的研究报告,发现22个CNVs可能与MR/DD相关。由于部分患儿同时携带MR/DD相关CNVs和未报道CNVs,故最终评估确认19例患儿具有MR/DD相关CNVs,1例患儿的CNVs临床意义不明确(表1)。因此,携带MR/DD致病相关CNVs的患儿检出率为17.1%(19/111例)。
19个与MR/DD相关的罕见CNVs中,1例为16p11.2微缺失综合征,Array-CGH结果见图2;2例为Pelizaeus-Merzbacher病;2例涉及15q11-13的Prader-Willi综合征/Angelman综合征(PWS/AS)核心区域,Array-CGH结果见图3;1例为Potocki-Lupski综合征,Array-CGH结果见图4;1例为Williams-Beuren综合征;1例为16p13.11微缺失综合征。
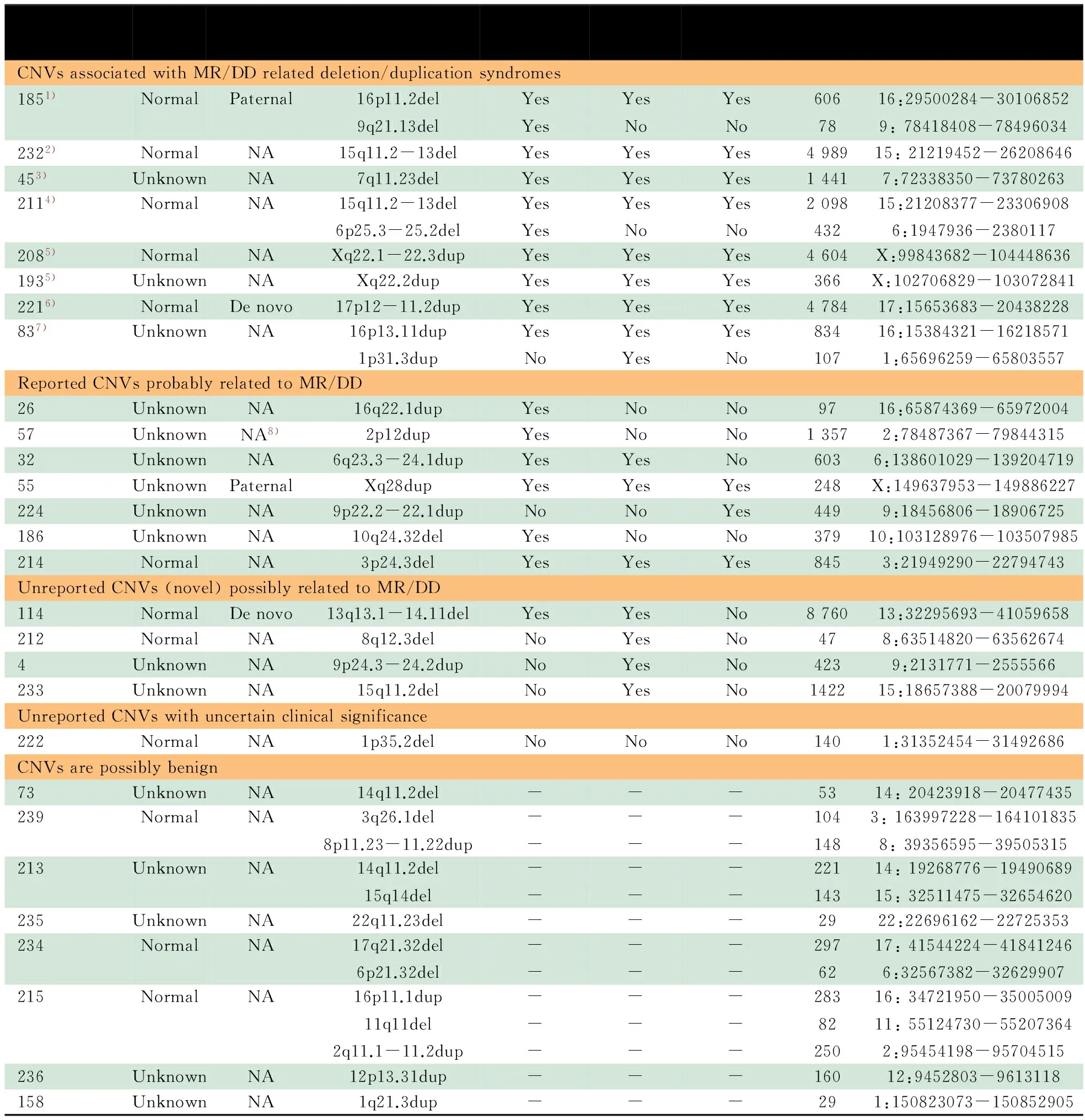
表1 不明原因MR/DD 患儿中CNVs类型特点
Notes 1) 16p11.2 Microdeletion syndrome; 2) Prader-Willi/Angelman syndromes (PWS/AS); 3) Williams-Beuren syndrome; 4) Atypical PWS; 5) Pelizaeus-Merzbacher disease;6) Potacki-Lupski syndrome; 7) 16p13.11 microdeletion syndrome; 8) father was NA, not maternal; NA: not available;-:not related

图2 16p11.2微缺失综合征患儿的Array-CGH 结果图
Fig 2 Array-CGH data of a patient with 16p11.2 deletion
Notes A showed a 606 kb deletion in 16p11.2 region from patient 185. Each probe was represented as a single dot and plots on X axis according to its genome position. Duplication/deletion was shown respectively as red/green dot and normal as black one. The green/red bar was represented for the affected region. B showed the distribution of Refseq gene in UCSC Browser (build 18) and known common CNVs in DGV in enlarged affected region. The bottom red bar represents for known common CNVs and upper scattered blue bar was represented for Refseq gene

图3 PWS患儿的Array-CGH 结果图
Fig 3 Array-CGH data of a patient with 15q11.2-13 deletion associated with PWS
Notes A showed an approximately 5 Mb deletion at 15q11.2-13 from patient 232. Each probe was represented as a single dot and plots on X axis according to its genome position. Duplication/deletion was shown respectively as red/green dot and normal as black one. The green/red bar was represented for the affected region. B showed the distribution of Refseq gene in UCSC Browser (build 18) and known common CNVs in DGV in enlarged affected region. The bottom red bar was represented for known common CNVs and upper scattered blue bar was represented for Refseq gene

图4 Potocki-Lupski综合征患儿的Array-CGH 结果图
Fig 4 Array-CGH data of a patient with Potocki-Lupski syndrome
Notes A showed an approximately 5 Mb duplication in 17p12-11.2 region from patient 221. Each probe was represented as a single dot and plots on X axis according to its genome position. Duplication/deletion was shown respectively as red/green dot and normal as black one. The green/red bar was represented for the affected region. B showed the distribution of Refseq gene in UCSC Browser (build 18) and known common CNVs in DGV in enlarged affected region. The bottom red bar was represented for known common CNVs and upper scattered blue bar was represented for Refseq gene
2.3 文献比对分析 在PubMed数据库通过文献检索,共获得26篇涉及MR的Array-CGH 或其他微阵列芯片研究的文献[2,7~31](包括MR人群研究和MR相关微阵列芯片研究回顾或综述)。通过比对共发现461个CNVs, 涉及312个不同染色体条带位点(图5),其中高发位点分别为22q11.2(19次),Xp22.31(16次),17p11.2(13次),1p36.33(10次),7q11.23(10次),17q21.31(10次),22q11.21(10次),Xq28(10次)。本研究发现的CNVs中, 11/36个(30.6%)曾被既往MR微阵列研究文献所报道。
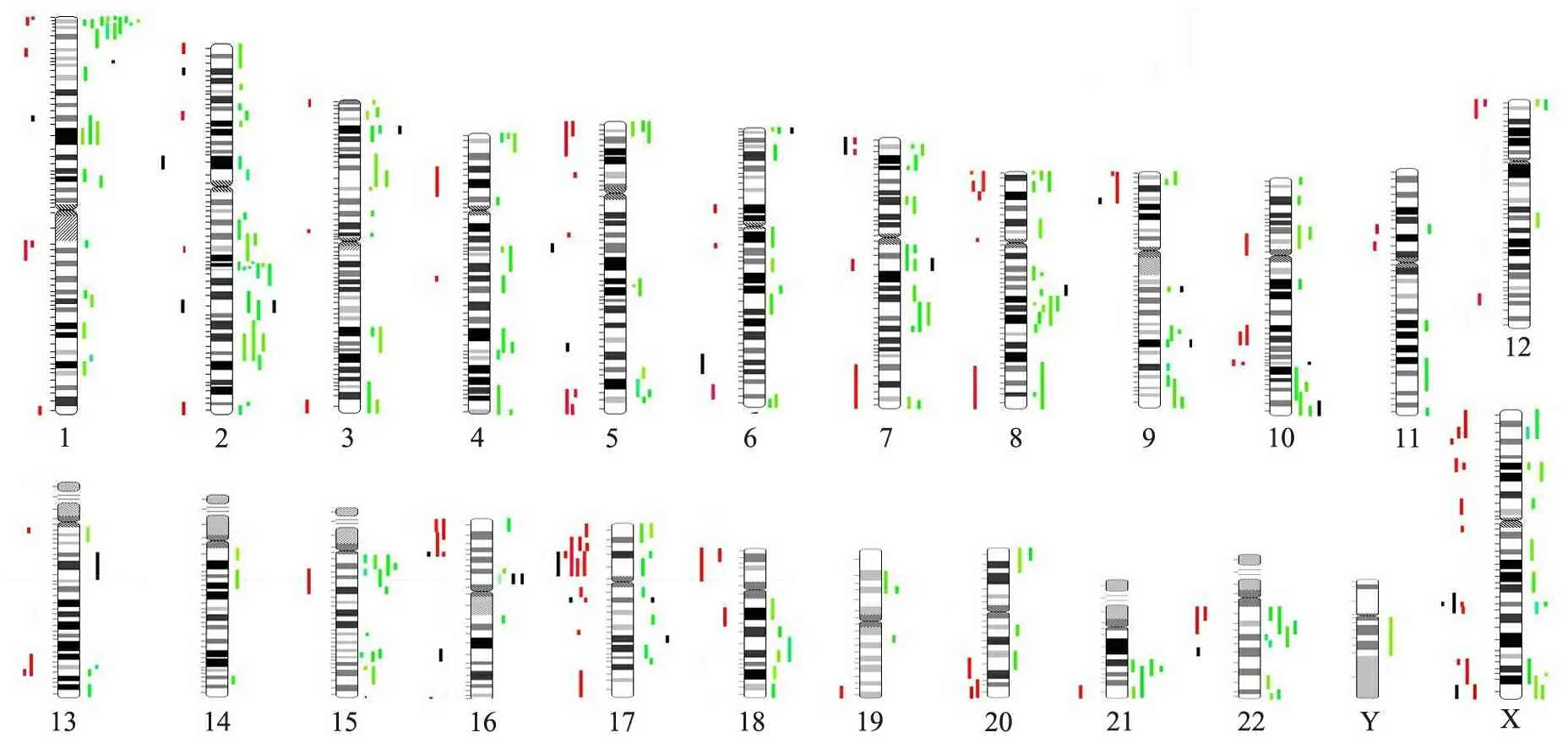
图5 MR相关CNVs在23条染色体上的分布
Fig 5 The distribution of CNVs related to MR on 23 chromosomes
Notes Rare CNVs from the literature review were mapped out according to position of 23 chromosomes (red/green). Red bar meant duplication and green meant deletion. Rare CNVs from our dataset are shown as black bars
2.4 1例非典型性PWS患儿的临床表型 病例号211,男,2岁3个月,G3P2,足月顺产,出生体重2 250 g,出生身长不详,生后喂养较困难,体重增长缓慢,9个月会抬头,1岁会坐,2岁会走,2岁会发“baba”和“mama”音,有惊厥史。父母否认近亲结婚、孕期药物毒物接触和家族性MR病史。查体:体重10.5 kg(<第1百分位,P1),身长80 cm( 图6 非典型性PWS患儿面部表型和MRI所见 Fig 6 The facial characteristics and MRI in a patient with atypical PWS Notes A,B:facial characteristics of patient 211 including flat face, sparse hair, hypertelorism and down-slanting eyes. Written consent to publish these images had been obtained from his legal guardian; C,D: MRI showed prominent bilateral frontal and temporal lobe sulcus, enlarged lateral ventricles 图7 非典型性PWS患儿的Array-CGH结果和Refseq基因图 Fig 7 The array-CGH data and scheme of Refseq gene in a patient with atypical PWS Notes A: Array-CGH data showed the deletion in PWS/AS critical region of patient 211;B: Scheme of chromosome band and involved refseq genes in 15q11-13 (21219452-26208646). In the top panel, an ideogram showed deletion band of proximal chromosome 15q11.2-13. Bottom panel showed affected genes in this region. In middle panel, smaller black bar was represented for deleted region in patient 211, bigger black bar was represented for patient 232. The common candidate genes of PWS/AS includeSNRPN,NECDIN,SnRNAs,UBE3A 3.1 Array-CGH在筛查MR/DD相关CNVs的诊断作用 Array-CGH属于DNA微阵列技术一种,是在原有高分辨染色体CGH基础上发展起来的分子遗传学技术,其基本原理是用不同的荧光染料分别标记待测和参考DNA样本,等量混合后与微阵列玻片上的oligonucleotide探针进行竞争性杂交。该技术通过一次杂交实验就可以获知整个基因组的CNVs,在染色体微结构改变、标记染色体来源的判定等方面具有明显的诊断优势,可检测出除基因突变或染色体平衡易位以外的几乎所有基因组失衡。 目前,欧美发达国家已将DNA微阵列技术应用于不明原因MR/DD的常规分子遗传检测,并进入到患儿医疗保险内。国外众多研究已证实在不明原因MR/DD患者中,10%~20%存在CNVs,并认为这种基因组失衡是MR/DD,甚至包括神经、精神疾患的致病原因之一[4,32,33]。另有遗传学专家提出可将微阵列芯片取代常规染色体检查,作为MR/DD患儿的首要检查项目,以减少患儿就诊费用和时间[31]。随着微阵列技术的不断发展,从BAC array,cDNA array到最新发展的寡核苷酸芯片(oligo array),从44 K到100 K再到244 K和1 M,微阵列芯片分辨率快速提高,即使50~100 K的微缺失或重复都能成功捕获,这使得MR/DD患儿基因组失衡的阳性检出率极大提高。而早期BAC 芯片,通常100 kb片段仅覆盖1个克隆,阳性检出率<10%[3]。本研究使用Agilent公司的Oligo 224 K芯片,阳性检出率为17.1%(19/111例),与国外研究报道大致相符[34],这些结果极大丰富了MR/DD的临床病因学研究,也说明该芯片可用于不明原因MR/DD的病因诊断。 除Array-CGH外,定量PCR、MLPA技术以及SNP array 也可对基因组进行拷贝数分析,定量PCR和MLPA技术由于涉及基因组特定位点,CNVs检出能力无法与微阵列芯片相比。国外研究显示,MLPA能发现5%~10%不明原因MR患儿存在亚端粒区域CNVs[35]。中国北京大学第一医院儿科曾利用MLPA分析39例MR患儿的23条染色体亚端粒结构,发现4例(10%)存在亚端粒拷贝数缺失或重复[5]。SNP array是另一种新发展的DNA微阵列,Affymetrix公司的SNP 6.0杂交芯片将SNP位点探针和已知CNVs探针结合,提高了分辨率和基因组CNVs的检出能力。Bernardini等[36]利用SNP 6.0芯片平台,重新对51个既往低分辨率芯片结果阴性的患者进行研究,显示6%患者存在3个可能致病的CNVs,且CNVs片段均>75 kb。此类芯片不仅可发现已知可能致病的CNVs,还可以分析杂合子丢失(loss of heterozygosity,LOH)和SNP位点信息。 3.2 MR/DD相关罕见CNVs的评估方法 随着比较基因组学研究的快速发展,如何发现和评估罕见致病性CNVs已成为一个面临挑战的重要任务。目前,国外很多分子诊断实验室都针对各自的实验平台,制定相应的评估方法[30,31,34,37],主要因素包括:①是否为新生CNVs;②是否在正常人群中有报道;③CNVs长度是否足够大;④是否在DECIPHER 或自己实验室的数据库中有报道,且携带者具有MR/DD表型[4]。但由于患儿双亲取样困难,因此临床信息、DGV和DECIPHER数据库比对显得尤为重要。本研究中将“是否曾在不明原因MR/DD人群中有报道”加入评估标准中,因为一些既往研究并没有将自己发现的罕见CNVs数据载入DECIPHER数据库。本研究结果显示,6个CNVs未被DECIPHER数据库报道。因此通过结合既往研究和美国波士顿儿童医院Array-CGH数据库,本研究最终将其中3个归为可能的致病性CNVs(表1),提高了MR/DD相关CNVs的诊断检出率,也提示既往文献回顾可作为MR/DD相关CNVs的一种评估方法。 3.3 非典型性PWS PWS/AS 是由于15q11-13区域上部分或全部印记基因簇发生突变、缺失、甲基化修饰异常或者单亲二体所引起。如果母本基因在该区域甲基化导致基因表达沉默而父本基因出现突变、缺失、甲基化异常或单亲二体则导致PWS,如果父本基因在该区域甲基化导致基因表达沉默而母本基因出现突变、缺失、甲基化异常或单亲二体则导致AS。15q11-13区域父本基因缺失是PWS最常见的病因[38];而母本基因异常则导致AS。本研究发现2例患儿(病例号232和211)存在PWS/AS关键区域缺失,其中1例(病例号232)缺失4 989 kb,涉及SNRPN、NECDIN、SnRNAs和UBE3A等重要印记基因簇,1例(病例号211)虽然缺失区域仅为2 098 kb,仍包含以上重要致病基因,从基因组特点可称为非典型性PWS。 本研究利用国际前沿的Array-CGH对中国人群中不明原因MR/DD患儿开展与MR/DD相关的CNVs研究,结果显示17.1%患儿携带可能与MR/DD相关的致病CNVs,其中已确定的部分罕见CNVs是中国人群中部分不明原因MR/DD患儿的发病原因之一。本研究同时发现,Array-CGH可帮助诊断某些与MR/DD相关但临床不易察觉的非典型性综合征,为部分不明原因的MR/DD患儿提供准确的遗传病因诊断。本研究作为转化医学研究的一项实践,不仅可将这个新的技术平台在中国及时推广应用于临床实践,加强和提高对不明原因MR/DD的分子诊断水平,而且对这些已发现的罕见CNVs区域所包括的许多基因可做进一步的挖掘,从中发现关键的致病主基因,并深入研究其致病机制,实现转化医学B2B(from bench to bed)的目标。 [1]Shevell M, Ashwal S, Donley D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society.Neurology,2003,60(3):367-380 [2]Shaffer LG,American College of Medical Genetics Professional Practice and Guidelines Committee. American College of Medical Genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation.Genet Med,2005,7(9):650-654 [3]Shaw-Smith C, Redon R, Rickman L, et al. Microarray based comparative genomic hybridization (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet,2004,41(4):241-248 [4]Vissers LE, de Vries BB, Veltman JA.Genomic microarrays in mental retardation: from CNV to gene, from research to diagnosis.J Med Genet, 2010 in press [5]Wu Y(吴晔),Jiang YW,Wang XZ,et al.Detection of subte-lomeric rearrangements in patients with idiopathic mental retardation/developmental delay.Chin J Pediatr(中华儿科杂志),2007,45(12):906-911 [6]Shen Y, Irons M, Miller DT, et al. Development of a focused oligonucleotide-array comparative genomic hybridization chip for clinical diagnosis of genomic imbalance. Clin Chem, 2007,53(12):2051-2059 [7]Vissers LE, de Vries BB, Osoegawa K, et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet,2003,73(6):1261-1270 [8]de Vries BB, Pfundt R, Leisink M, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet,2005,77(4):606-616 [9]Schoumans J, Ruivenkamp C, Holmberg E, et al. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH). J Med Genet,2005,42(9):699-705 [10]Menten B, Maas N, Thienpont B, et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet,2006,43(8):625-633 [11]Rosenberg C, Knijnenburg J, Bakker E, et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: clinical significance of imbalances present both in affected children and normal parents. J Med Genet,2006,43(2):180-186 [12]Sharp AJ, Hansen S, Selzer RR, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet,2006,3(9):1038-1042 [13]Aradhya S, Manning MA, Splendore A, et al. Whole-genome array-CGH identifies novel contiguous gene deletions and duplications associated with developmental delay, mental retardation, and dysmorphic features. Am J Med Genet A,2007,143A(13):1431-1441 [14]Baris HN, Tan WH, Kimonis VE, et al. Diagnostic utility of array-based comparative genomic hybridization in a clinical setting. Am J Med Genet A,2007,143(21):2523-2533 [15]Engels H, Brockschmidt A, Hoischen A, et al. DNA micro-array analysis identifies candidate regions and genes in unexplained mental retardation. Neurology,2007,68(10):743-750 [16]Fan YS, Jayakar P, Zhu H, et al. Detection of pathogenic gene copy number variations in patients with mental retardation by genomewide oligonucleotide array comparative genomic hybridi-zation. Hum Mutat,2007,28(11):1124-1132 [17]Lu X, Shaw CA, Patel A, et al.Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases.PLoS One, 2007,2(3):e327 [18]Shaffer LG, Bejjani BA, Torchia B, et al. The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. Am J Med Genet C Semin Med Genet, 2007,145C(4):335-345 [19]Thuresson AC, Bondeson ML, Edeby C, et al. Whole-genome array-CGH for detection of submicroscopic chromosomal imbalances in children with mental retardation. Cytogenet Genome Res, 2007,118(1):1-7 [20]Wagenstaller J, Spranger S, Lorenz-Depiereux B,et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet, 2007,81(4):768-779 [21]Pickering DL, Eudy JD, Olney AH, et al. Array-based comparative genomic hybridization analysis of 1176 consecutive clinical genetics investigations. Genet Med,2008,10(4):262-266 [22]Lybaek H, Meza-Zepeda LA, Kresse SH, et al. Array-CGH fine mapping of minor and cryptic HR-CGH detected genomic imbalances in 80 out of 590 patients with abnormal development. Eur J Hum Genet, 2008,16(11):1318-1328 [23]Nowakowska B, Stankiewicz P, Obersztyn E, et al. Application of metaphase HR-CGH and targeted Chromosomal Microarray Analyses to genomic characterization of 116 patients with mental retardation and dysmorphic features. Am J Med Genet A,2008,146A(18):2361-2369 [24]Krepischi-Santos AC, Vianna-Morgante AM, Jehee FS, et al. Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromes revisited and new alterations. Cytogenet Genome Res,2006,115(3-4):254-261 [25]Friedman JM, Baross A, Delaney AD, et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet, 2006, 79(3):500-513 [26]Koolen DA, Vissers LE, Pfundt R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet,2006,38(9):999-1001 [27]Lugtenberg D, de Brouwer AP, Kleefstra T, et al. Chromosomal copy number changes in patients with non-syndromic X linked mental retardation detected by array CGH. J Med Genet,2006,43(4):362-370 [28]Shevell MI, Bejjani BA, Srour M, et al. Array comparative genomic hybridization in global developmental delay. Am J Med Genet B Neuropsychiatr Genet,2008,147B(7):1101-1108 [29]Sagoo GS, Butterworth AS, Sanderson S, et al. Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genet Med,2009,11(3):139-146 [30]McMullan DJ, Bonin M, Hehir-Kwa JY, et al. Molecular karyotyping of patients with unexplained mental retardation by SNP arrays: a multicenter study. Hum Mutat,2009,30(7):1082-1092 [31]Gijsbers AC, Lew JY, Bosch CA,et al. A new diagnostic workflow for patients with mental retardation and/or multiple congenital abnormalities: test arrays first. Eur J Hum Genet, 2009,17(11):1394-1402 [32]St Clair D.Copy number variation and schizophrenia.Schizophr Bull,2009,35(1):9-12 [33]Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med,2008, 358(7):667-675 [34]Hochstenbach R, van Binsbergen E, Engelen J,et al.Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands.Eur J Med Genet,2009,52(4):161-169 [35]Koolen DA,Nillesen WM,Versteeg MH,et al.Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA).J Med Genet,2004,41(12):892-899 [36]Bernardini L, Alesi V, Loddo S,et al.High-resolution SNP arrays in mental retardation diagnostics: how much do we gain?Eur J Hum Genet, 2010,18(2):178-185 [37]Koolen DA, Pfundt R, de Leeuw N, et al. Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum Mutat,2009,30(3):283-292 [38]Prader-Willi syndrome association.http://www.pwsausa.org/syndrome/index.htm

3 讨论
4 总结
猜你喜欢
昆明医科大学学报(2022年4期)2022-05-23
昆明医科大学学报(2021年12期)2021-12-30
今日农业(2021年11期)2021-08-13
现代临床医学(2021年2期)2021-03-29
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
中成药(2018年7期)2018-08-04
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
