
改性活性炭吸附氮化物的机理研究
2010-01-12 12:24:34解治香胡廷平
武汉轻工大学学报 2010年4期
解治香,胡廷平
(武汉工业学院化学与环境工程学院,湖北武汉 430023)
随着环保意识增强,人们对燃油的使用标准制定了严格要求,同时,燃油的普及使得人们必须对燃油纯化,包括金属、硫、氮等杂质的脱除。脱氮是近年来随着环保意识增强开始研究的,研究表明燃油精制前脱氮明显提高脱硫深度,降低加氢操作条件[1-3],同时减少环境污染。
燃油中氮化物含量随产地不同而不同,一般少于硫含量[4],但是氮化物会阻碍 HDS的深度。碱性氮化物吸附催化剂上阻碍硫化物的脱除而非碱性氮化物在 HDS过程中转化为碱性氮化物,而且氮化物的存在还会造成油品安定性降低,颜色变深,腐蚀金属设备,在燃烧中生成 NOx造成大气污染[5-7]。氮化物大体上分为碱性氮化物和非碱性氮化物两类。碱性氮化物以吡啶、喹啉及其衍生物为主,非碱性氮化物以吡咯、吲哚及衍生物为主[8]。本文针对油品中有机芳香氮化物的难脱问题,从微观方面利用分子模拟软件对脱氮机理进行研究。
1 实验部分
1.1 计算理论
本研究基于量子力学方法、分子力学方法和蒙地卡罗方法进行实验[9]。量子力学是以分子中电子的非定域化为基础,电子行为以其波函数表示,包括三种近似方法:从头计算法 (abinition method),半经验法 (semi-empirical molecular orbital method)和密度泛函数理论 (density functional theory)。分子力学是根据经典力学的计算方法,该方法主要依据分子力场计算分子的各种特性,广泛应用于计算分子的构象和能量。分子力场的许多参数由量子力学计算或实验方法等得到。蒙地卡罗计算法借由系统中质点的随机运动,结合统计力学的概率分配原理,得到体系的统计和热力学资料。
1.2 计算方法
本文以吡啶、喹啉和 2-甲基喹啉为碱性氮化物,以吡咯、吲哚和 2-甲基吲哚为非碱性氮化物。利用Materials Studio 4.4软件中的 Dmol3模块,采用密度函数 (DFT),对碱性氮化物和非碱性氮化物几何构型优化,函数采用广义梯度 (GGA)的 PW91,基组为DND,收敛精度为程序设定值。草酸亚铁通过 Forcite几何优化,获得稳定几何构型。活性炭表面上的草酸亚铁是利用 Visualizer模块对优化后的草酸亚铁表面定位,而后利用 Forcite模块能量优化。改性活性炭与氮化物模拟吸附利用 Adsorption Locator模块,采用退火方式模拟吸附。
2 结果与讨论
2.1 碱性氮化物和非碱性氮化物的结构
各图形所代表的元素符号:


表1 碱性氮化物和非碱性氮化物优化后的结构
表1是碱性氮化物与非碱性氮化物通过 Dmol3模块优化后的结构,喹啉、2-甲基喹啉、吲哚和 2-甲基吲哚分子分别比吡啶和吡咯的分子多一个苯环,同时碱性氮化物含N环是六元环,N上不含有 H,N上含有一对孤对电子,显碱性;而非碱性氮化物含 N环是五元环,N上有 H,N上不含有孤对电子,显中性。
它们最高占据分子轨道 (HOMO)和最低未占据分子轨道能量 (LUMO)如表2。

表2 碱性氮化物和非碱性氮化物的轨道
从表2知碱性氮化物的最高占据分子轨道能量比非碱性氮化物低,最低未占据分子轨道的能量比非碱性氮化物的低,易于反应,易于从油品中脱除。
2.2 草酸亚铁分子结构
草酸亚铁的结构如图1所示。
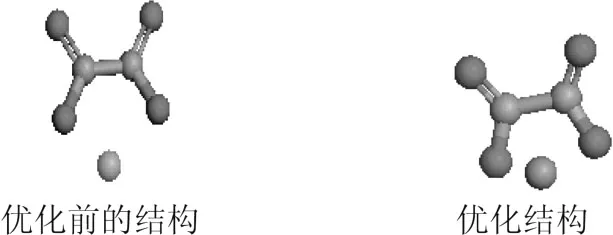
图1 优化前后草酸亚铁结构
2.3 改性活性炭吸附碱性氮化物
表3是改性活性炭吸附碱性氮化物的构型,喹啉与吡啶的最小吸附能分别为 -130.03 kcal/mol,-120.70 kcal/mol,相差不大。但吸附构型的能量喹啉为 -58.13 kcal/mol而吡啶为 -107.32 kcal/mol,相差近 50 kcal/mol。喹啉有一苯环,在吸附过程中起到位阻作用。从表3知脱除碱性氮化物主要是N上的孤对电子起作用,它与铁原子的 S空轨道结合,形成酸碱络合物。构型 1就是这种方式,吸附能最小,总能量最小,容易结合,结构相对稳定。构型 2是含 N环与草酸亚铁中的铁作用,铁原子 d轨道电子与含 N环空π*分子轨道结合。构型 3是苯环空π*轨道与铁原子 d轨道电子结合。2-甲基喹啉由于氮原子邻位上的甲基和苯环的位阻作用,极大限制了氮原子的接触范围。2-甲基喹啉的吸附能与吡啶和喹啉相差 100 kcal/mol以上,并且它的构型能为正值。甲基的存在使得 2-甲基喹啉酸碱络合以构型 1和构型 4两种方式接触。由表3可知,碱性氮的脱除主要以酸碱络合为主,π轨道结合为辅。

表3 改性活性炭吸附碱性氮化物的构型
2.4 改性活性炭吸附非碱性氮化物
由表4可知 2-甲基吲哚的吸附能仅为负十几,而吡咯与吲哚的最小吸附能却为 -97.87 kcal/mol,-101.75 kcal/mol,相差很大;而吸附构型的能量各为-69.45 kcal/mol,50.55 kcal/mol,110.90 kcal/mol,吲哚和 2-甲基吲哚相差不大,能量较高,不稳定,主要由于苯环与甲基的位阻作用。吸附非碱性氮化物有两种作用,一是π络合,含 N环空的π*分子轨道与铁原子 d轨道电子结合,这种构型吸附能量较低,非碱性氮主要以这种形式络合脱除。吡咯构型 1、吲哚构型 1、吲哚构型 2和 2-甲基吲哚构型 1属于这种络合形式。吲哚构型 3和 2-甲基吲哚构型 2是苯环π*空轨道与铁原子 d轨道的电子结合;二是形成 H键,非碱性氮化物由于 N的极性较大,N与 H形成σ键的电子对偏向 N原子,H几乎成为裸原子,因此易于形成 H键。吡咯构型 2和 3,吲哚构型 5和 2-甲基吲哚构型 3,4,5是与草酸亚铁中羰基上的氧形成 H键,吡咯构型 4和吲哚构型 4是与碳氧单键上的氧形成 H键。吡咯形成 H键构型中,由于空间位阻使得羰基氧的 H键构型反而比碳氧单键氧的 H键构型能量低 30 kcal/mol左右;由于本身苯环位阻的影响,吲哚形成 H键构型的能量没有多大区别。而 2-甲基吲哚由于甲基和苯环的影响使得 H键结合与π络合的吸附能相差不大,受甲基影响,2-甲基吲哚从草酸亚铁的背面以 H键结合,吸附能为 -16.66 kcal/mol,比π络合吸附能 -15.86 kcal/mol还要小。可见随着位阻的增加,H键结合相对π络合更容易。

表4 改性活性炭吸附非碱性氮化物的构型
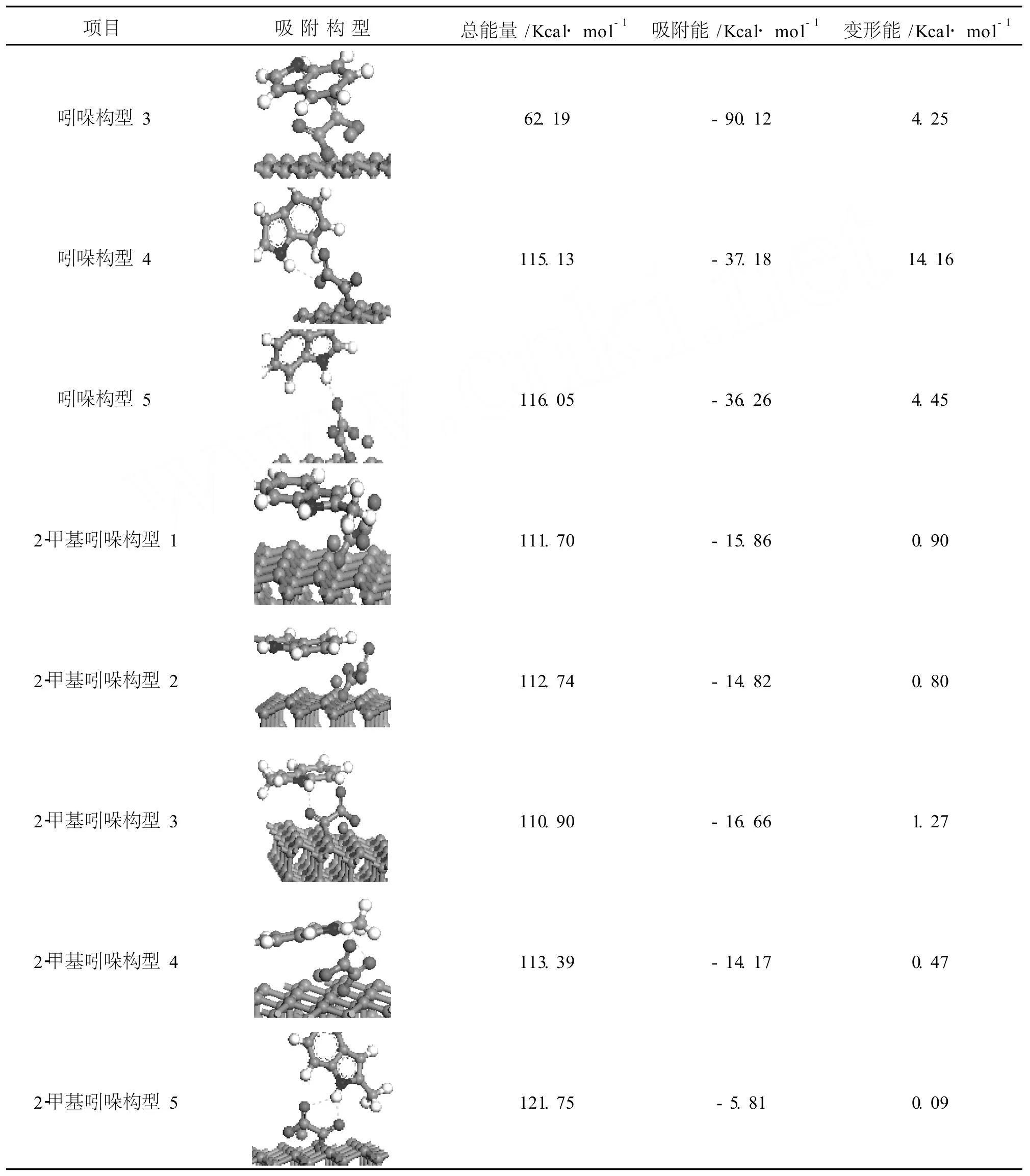
续表
2.5 改性活性炭吸附位点
改性活性炭吸附氮化物位点如图2示。图2中上部深色区代表氮化物吸附位点的几率。从图2可看到,改性活性炭起到吸附作用点主要在铁原子周围。

图2 改性活性炭吸附氮化物位点
2.6 改性活性炭吸附碱性氮化物和非碱性氮化物的比较
碱性氮化物与非碱性氮化物的吸附能和变形性相差不大,主要在于整个络合物分子能量上,整个碱性络合物分子的能量比较小,构型稳定,而非碱性氮化物整个分子能量比较高,尤其是吲哚构型的能量,它们三种物质构型能都为负值,而吲哚是正值且较大,稳定性差。因此,碱性氮化物比非碱性氮化物易于脱除。
3 结论
通过利用分子模拟软件对碱性氮化物和中性氮化物的吸附行为进行模拟,我们得出碱性氮化物的吸附主要以酸碱络合为主;而非碱性氮化物以形成π-络合物为主,H键结合为辅。在以后脱氮研究工作中对碱性氮化物从酸碱络合方向研究;对非碱性氮化物从形成π-络合物和 H键方向研究。
[1] Liu ZL,Zhang Q K,Zheng Y,et al.Effect of nitrogen and aromatics on hydrodesulfurization of light cycle oil predicted by a system dynamics model[J].Energy&Fuels,2008,22:860-866.
[2] Jayaraman A,Yang F H,Yang R T.Effect of nitrogen compounds and polyaromatic hydrocarbons on desulfurization of liquid Fuels by adsorption via π-Complexatiion with Cu(Ⅰ)Y zeolite[J]. Energy&Fuels,2006,20:909-914.
[3] Sano Y S,Choi K H,Korai Y,et al.Effects of nitrogen and refractory sulfur species removal on the deep HDS of gas oil[J].Applied Catalysis,2004,53:169-174.
[4] 陈绍洲.石油化学[M].上海:华东理工大学出版社,1993.
[5] Yang H,Chen J W,Briker Y G,et al.Effect of nitrogen removal from light cycle oil on the Hydrodesulphurization of dibenzothiophene,4-methyldibenzothiophene and 4, 6-dimethyldibenzothiophene[J].Catalysis Today,2005,109:16-23.
[6] Choi K H,Korai Y,Mochida I,et al. Impact of removal extent of nitrogen species in gas oil on its HDS Performance:an efficient approach to its ultra deep desulfurization [J]. Applied Catalysis,2004,50:9-16.
[7] Almarri M,Ma X L,Song C S. Selective adsorption for removal of nitrogen compounds from liquid hydrocarbon stream over carbon-and alumina-based adsorbents[J]. Indusrial&Engineering Chemistry Research,2009,48:951-960.
[8] 吕志凤,战风涛,李林,等.催化裂化柴油中氮化物的分析 [J].石油化工,2001,30(5):399-401.
[9] 陈正隆,徐为人,汤立达,等.分子模拟的理论与实践[M].北京:化学工业出版社,2007.
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13 06:40:42
石油炼制与化工(2021年5期)2021-05-12 06:38:46
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
航空制造技术(2020年15期)2020-11-06 09:10:38
表面技术(2019年6期)2019-06-26 10:20:34
山东化工(2019年11期)2019-06-26 03:26:44
国际呼吸杂志(2019年1期)2019-03-08 03:07:02
中成药(2017年7期)2017-11-22 07:33:25
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
合成化学(2015年1期)2016-01-17 08:59:30
