
酿酒酵母2B-39 sam2在大肠杆菌中的克隆及表达
2008-04-26 03:32安胜欣江章应彭成松
安徽理工大学学报·自然科学版 2008年3期
关键词:克隆
安胜欣 江章应 彭成松
摘 要: 构建S-腺苷甲硫氨酸合成酶基因(sam2)的基因工程菌,为S-腺苷甲硫氨酸(S-ade nosylmethionine,SAM)的工业化生产提供参考。应用PCR技术从酿酒酵母的总DNA中扩增出 1.2 kb的sam2,构建重组载体pET28a-sam2,将其转入大肠杆菌进行表 达和分析。SDS-PAGE显 示重组克隆表达的SAM2分子量约47 kD,重组蛋白量约细菌总蛋白的20.1% ,酶活达到19.6 nmol/(h•mL),大肠杆菌表达出了具有生物活性的sam2。
关键词:酿酒酵母;SAM;克隆
中图分类号:Q786 文献标识码:A 文章编号:1672-1098(2008)03-0065-04
SAM是生物体内一种重要的生理活性物质,参与体内40多种生化反应[1-4],对关 节 炎、抑郁症、肝功能紊乱等均有较好的疗效,而且还是预防癌症、心血管疾病和抗衰老的高 级保健品[5]。因此,研制、开发SAM具有重要理论和实际意义。
尽管SAM的疗效已得到人们的认可,但生产成本昂贵使其应用受到限制。目前国内外都 在竞相开发廉价的SAM合成工艺。SAM的制备主要有发酵法和酶促合成法[6-8], 后者虽具有终产物积累量高,易分离纯化等优点,但因原料ATP成本高,已很少使用。发酵 法因其生产 工艺简单、成本低成为较为有效的工业化生产方法,但受微生物自身条件的限制,如SAM 合成酶含量少、酶活不高[9],而且分离纯化困难。随着基因工程技术的发展,利 用高活力的腺苷甲硫氨酸合成酶基因工程菌发酵生产SAM以提高其产量成为可能。而酿酒酵 母sam2的活力较其它微生物高成为理想的酶源[10]。就目前已经构建的基因工 程菌[11-13]来说,大肠杆菌具有生产周期短、工艺操作简单、原料成本低等优点 ,是较为理想的宿主来源。本文以酿酒酵母作为出发菌株,克隆sam2,并将其转入大肠 杆菌中进行表达,旨在为SAM的廉价工业化生产提供参考。
1 材料和方法
1.1 材料
酿酒酵母、E.coli DH5 a、E.coli BL21(DE3)均为实验室保存;pUCm-T 载体表 达载体p ET28a购自上海生物工程有限公司。各种限制性内切酶、Taq DNA聚合酶、T4 DNA连接酶及DN A ladder、各种试剂盒均为TaKaRa公司产品;IPTG、氨苄青霉素、卡那霉素为Merck公司产 品;低分子量蛋白质标准为上海生物化学研究所产品;丙烯酰胺,2N′-亚甲基双丙烯酰胺 为欣经科公司产品;SAM标准品为Sigma公司产品;其他生化试剂或常规试剂均为分析纯或者 超纯。
1.2 方法
(1) 酿酒酵母基因组DNA的提取方法 按照基因组DNA提取试剂盒说明 书进行制备。
(2) PCR扩增sam2 参考GenBank中(登录号M23368)sam2的 基因序列设计软件PCR引物:
上游引物P1: 5′-CGGAATTCATGTCCAAGAGCAAAACTTT-3′
下游引物P2: 5′-CCAAGCTTAGCATAAAGAAAGGGATTGA-3′
其中上、下游引物中加入EcoR I、Hind III酶切位点。 以1.2.1提取的基 因组DNA为 模板,以P1、 P2为引物扩增sam2。 PCR反应参数为: 模板 2 μL; 10× 缓冲液 5 μL;引物1、引物2(20 μM) 2 μL;四种 脱氧 核苷酸(25 mM) 0.5 μL; MgCl2(25 mM) 3 μL;DNA聚合酶0.5 μL;总体积50 μL。PCR反应条件: 9 4 ℃ 变性5 min; 94 ℃ 30 s, 55 ℃复性45 s、 72 ℃延伸1 min, 30个循环;最后72 ℃延伸10 min。PCR产物在1%的琼脂糖凝胶上电泳。
(3) 重组质粒的构建及鉴定 用DNA快速纯化片断回收试剂盒回收PCR产物中约1 .2 kb的DNA 片断。回收片断与载体pUCm-T连接过夜后,转化E.coli DH5 a,菌液涂布于含有IPTG 、X-gal及氨苄青霉素(70 μg/μL)LB平板上,37 ℃培养24 h后将长有菌落的平板置4 ℃冰箱数小时,用消毒的牙签挑取 白色菌落,提取质粒,EcoR I、Xhol I双酶切鉴定筛选阳性重组子。将筛选 的阳性重组子pUCm-sam 2送上海生物工程公司测序分析。将鉴定正确的重组质粒pUCm-sam2切下目的片断后,与 用此两种酶双酶切的表达载体pET28a连接,构建重组表达质粒pET-sam2,转入E.coliBL21(DE3),涂布于含卡那霉素(30 μg/μL)的LB平板上,酶切鉴定以筛 选阳性。
(4) sam2的诱导表达及SDS-PAGE分析 将鉴定的阳性克隆单菌落 3 7 ℃培养过夜后以1%接种量接种于含卡那霉素(30 μg/μL)的LB 培养 基中扩大培养,继续培养至A600=0.5时,添加IPTG至终浓度为1 mmo l/L 来诱导表达SAM2。对诱导表达的不同时间取样进行分析。样品离心收集菌体,用100 μL的上样缓冲液重悬。沸水煮5 min,高速离心,取10 μL 上清按常规方法进行SDS-PAGE(分离胶质量分数为12%)电泳。
(5) sam2表达产物的鉴定及酶活测定 将转化菌扩大培养到10 0 mL LB培养基中, 37 ℃, IPTG 1 mmol/L诱导表达 一定时 间后离心收集菌体, 用PBS缓冲液洗2次,加溶菌酶至质量浓度为1.5 mg/L,3 7 ℃缓慢振荡3 h,用0.2 M的HCl调节pH至4.5,镜检的 细胞破碎情况,离心收集细胞碎片(4 ℃,12 000 g,20 min)和上清液。将细胞碎片和上清液进行SDS-PAGE凝胶分析。
酶活力参考文献[6]296进行测定。酶的活力单位定义为:在上述反应条件下,37 ℃,60 min催化形成1 μmolSAM所需要的酶量为一个 活力单位。
2 结果
2.1 sam2的扩增
将PCR产物在1.0%的琼脂糖凝胶上电泳,可得到一条约1.2 kb的DNA片断,与s am2基因编码的序列大小一致(见图1)。
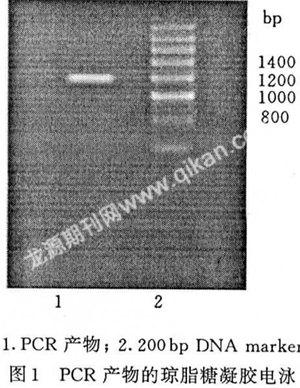
2.2 重组质粒的鉴定
随机挑取转化平板上的白色菌落,37 ℃振荡培养后快速提取质粒,将经双酶切 鉴定筛选的阳性重组子pUCm-sam2送至上海生工测序鉴定,测序结果表明有四个核苷酸 发生突变,但由于密码子的简并性,氨基酸并没有发生改变。将上述sam2基因片段与pE T28a连接后,转入E.coli BL21(DE3),涂布于含卡那霉素(30 μg/μL) 的L B平板上,37 ℃过夜培养。随机挑选菌落,抽提质粒进行限制性内切酶酶切鉴 定,经1.0%的琼脂糖凝胶电泳分析,分析结果表明pET28a上也插入了1.2 kb的D NA片断见图2。

2.3 sam2的SDS-PAGE
SDS-PAGE检测结果如图3, IPTG诱导1 h, 2 h,4 h ,6 h,8 h,10 h,外源蛋白S-腺苷甲 硫氨酸合成酶均有表达,且随着时间的延长表达量增大。对电泳结果扫描显示,在8 h时表达量最大,可达到细菌总蛋白20.1%。外源蛋白的相对分子量约为47 kD,其中包含了利用该载体在重组蛋白N端附加的34个氨基酸的相对分子量,附加的氨基 酸含有6个组氨酸的标记蛋白。所测得的相对分子量减去载体自带的34个氨基酸的相对分子 量与酿酒酵母中sam2表达的蛋白的相对分子量43 kD是相符的。
2.4 sam2表达产物的鉴定及酶活测定
将经1.2(5)处理后的上清和细胞碎片进行SDS-PAGE凝胶分析(见图4)。由图4可以看出 sa m2表达蛋白大多存在于细胞碎片中,以包涵体形式存在,以水溶形式存在的量很少。
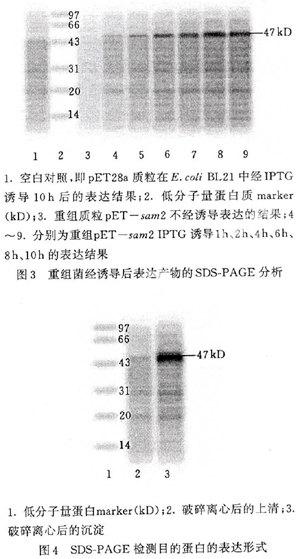
图4 SDS-PAGE检测目的蛋白的表达形式 将上述处理得到包涵体用洗涤液洗涤,然后溶解在含8 mol/L的尿素变性缓冲液 中,4 ℃放置过夜后稀释20倍,加入还原型谷胱甘肽(2 mmol/L) 和氧化型谷胱甘肽(0.5 mmol/L)于4 ℃复性24 h。 最后用0.45 μm膜过滤后, 用镍离子亲和层析浓缩纯化重组蛋白。 用纯化后 的蛋 白测定酶活力。 高效液相色谱测定结果表明: 纯化蛋白的酶活达到19.6 nmol /(h•mL), 大肠杆菌表达出有活性的S-腺苷甲硫氨酸合成酶。
3 讨论
大肠杆菌是基因工程中采用最多的蛋白质表达体系,因其遗传背景清楚、技术操作简便 、培养条件简单、大规模发酵经济,常常作为外源基因高效表达的首选体系。但是因大肠杆 菌结构简单,细胞缺少真核细胞的转录后加工系统,并且作为大肠杆菌原核表达载体不具备 识别内含子、外显子的能力。根据酿酒酵母的蛋白数据库及其基因组数据库的信息表明,酿 酒酵母的sam2不具备内含子,所以可以直接以酿酒酵母的染色体DNA为模板,为工业化 生产奠定基础。
本文通过一系列的分子生物学操作,成功的构建了重组表达载体pET-sam2并将其转入大 肠杆菌BL21(DE3)中进行了诱导表达。SDS-PAGE图显示外源蛋白分子量的大小约为47 kD,而理论值为43 kD,是除了sam2基因得以正常表达外,表达载体 自身的102个碱基也得到了表达。外源蛋白减去表达载体上的102个碱基所翻译的34个氨基酸 的相对分子量后大约为43 kD,与理论相符。
基因表达产物能否在宿主细胞中稳定积累而不被内源蛋白水解酶所水解是克隆基因的有 效表达的一个重要因素。某些外源蛋白可以在宿主细胞中以包涵体的形式表达,这种不溶性 的沉淀复合物可以抵抗宿主细胞蛋白酶的降解,也便于纯化。然而经包涵体纯化的重组蛋白 必须经过变性-复性的处理,才能获得具有生物活性的蛋白。但在变性-复性的处理中可能对 蛋白质的天然构象造成不可预知的影响[14]。有本实验结果,sam2酶活力为19 .6 nmol/(h•mL)可以看出,酶活力相对较低,这可能就是在变性-复性的处理 中,其天然构象发生变化,使 活性降低;另外,包涵体是也是蛋白高效表达的结果,低温培养可以防止包涵体的生成,以 后实验可以在低温条件下进行,避免形成包涵体影响酶活力。也可以通过优化发酵条件,提 高sam2的酶活以提高蛋白的产量。
参考文献:
[1] 余志良,杨晟,蔡谨.S-腺苷甲硫氨酸研究进展[J].2003,34(1):49-53.
[2] KONIG B.A long-term(two years) clinical trail with S-adenosy l-methionine for the treatment of osteoarthritis[J].Am J Med,1987,83(suppl 5 A):89-69.
[3] BITTIGLIERI T.Admetionie(S-adenosyl-methionine)neurologicology:Implication for drug therapies in psychiatric and neurological disorders[J]. E xpert opin Invest Durgs,1997,6(4):417-430.
[4] MATO J M,ALBAREZ L,ORTIZ P, et al. S-ade-
nosyl-L-methionine syn th etase and methionine metabolism deficiencies in cirthosis[J].Adex Exp Med,1994 ,368:113-119.
[5] 汤亚杰,李艳等,S-腺苷甲硫氨酸的研究进展[J].生物技术通报,200 7, 3: 76-81.
[6] LI DY,YU J,JI XS,et al.Production of SAM by recombinant Pichia pas toris[J].J Biotechnol, 2002,18(3):295-299.
[7] MATOS JR, RAUSHEL FM, WONG CH. S-ade-
nosylmethionine: Studies o n che mical and enzymatic synthsis[J].Biotechnol Appl Biochem,1987,9(1):39-52.
[8] 公剑,韦平和.S-腺苷甲硫氨酸合成酶及其在S-腺苷甲硫氨酸合成在中 的应用[J].药物生物技术,2001,8(2):108-111.
[9] SHIOZAKI S,SHIMIZU S,YAMADA H.Unusual intracellular accumul ation of S-adenosyl-L-methionine by microorganisms[J].Agric Biol Chem,1984,48 (9):2 293-2 300.
[10] TSUTOMU KODAKIL,HINJI TSUJIL, NAOKO
OTANI.Differential tran scriptional reg ulation of two distinct S-adenosylmethionine synthetase gene(sam1 and sam2 )o f Saccharomyces cerevisiae[J].Nucl Acids Res Suppl, 2003,3:303-304.
[11] 张建国,蔡瑞,李新华.发酵重组Pichia pastoris生产腺苷甲硫氨酸的研 究[J].工业微生物,2004,34(4):1-5.
[12] 陶敏,干信.S-腺苷甲硫氨酸合成酶在大肠杆菌中的表达与克隆[J].生物 技术,2006,16(3):20-22.
[13] 吴雪昌,应晓屹.提高酵母S-腺苷甲硫氨酸表达量的研究[J].中国药学杂 志,2007,42(8):564-571.
[14] 姚文斌.生物技术制药概论[M].北京:中国医药出版社,2006:53-58.
(责任编辑:李 丽)
猜你喜欢
环球时报(2022-09-20)2022-09-20
今日农业(2020年24期)2020-12-15
河北果树(2020年2期)2020-05-25
小学科学(学生版)(2019年5期)2019-05-21
中央民族大学学报(自然科学版)(2018年1期)2018-06-27
兽医导刊(2016年12期)2016-05-17
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
水生生物学报(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
