
基于mtDNA D-loop的河南某地引入猕猴群体遗传背景分析
2025-02-21 00:00:00周言言王钰炜田军东路纪琪
野生动物学报 2025年1期


关键词:猕猴;物种引入;种群;遗传多样性;系统发育
野生动物旅游是以野生动物为主要消费对象的一种生态旅游活动,而猕猴(Macaca mulatta)则是最为常见的目标动物之一[1]。自20世纪80年代中期以来,基于野生动物的生态旅游快速发展,并取得了可观的经济利益[2]。在此背景下,一些地区通过引入(introduction)或重引入(re-introduction)方式,使猕猴(或其他野生动物)扩散到其自然分布区以外的地方[3]。然而,引入来源不清、遗传背景不明的动物,可能导致不同进化谱系之间产生遗传同质化、遗传污染等现象,进而扰动局部地区的动物区系组成,甚至引发严重的生态后果[4]。如棕树蛇(Boiga ir⁃regularis)[5]、褐家鼠(Rattus norvegicus)[6]和温室蟾(Eleutherodactylus planirostris)[7−8]等动物,借由交通运输和贸易等人类介导的方式扩散到新的地区。被引入物种一旦定殖成功,其种群数量往往迅速增长[9−11],并通过竞争、捕食和疾病传播等方式,严重威胁本土物种与生态系统稳定,最终成为入侵物种。美洲牛蛙(Rana catesbeiana)导致全球多个入侵地区的本土两栖类快速下降甚至灭绝[12];缅甸蟒(Pythonbivittatus)入侵北美地区后,造成当地小型兽类种群的快速下降、濒临灭绝[13];褐家鼠等入侵鼠类导致鼠疫、鼠伤寒在全球范围内传播[14]等。
遗传多样性常用于评估物种对环境的适应能力和抵御疾病的能力[15−18]。在物种灭绝后的重引入过程中,遗传多样性水平被作为重要的参考指标[16,19−20]。如对麋鹿(Elaphurus davidianus)[21−22]、普氏野马(Equus ferus przewalskii)[23]和大熊猫(Ailu⁃ropoda melanoleuca)[24]等物种的研究结果,无不展示了遗传多样性在珍稀濒危物种保护中的重要作用。较高的遗传多样性有助于物种快速响应环境变化、减少近交,也是入侵物种成功定殖的主因[25−27]。因此,评估引入物种的遗传多样性水平、追溯其原始产地,对建立规范、可行和有效的监管措施极为必要[28]。
猕猴是世界上分布最广泛的非人灵长类物种[29−30],在我国的分布范围从热带雨林到温带雪山[31−32]。笔者在野外调查期间,于河南省荥阳市环翠峪风景名胜区(34°64′ N,113°27′ E)发现一群自由活动的引入猕猴群体,遂予以重点关注。迄今为止,关于该猕猴群体的数量现状、遗传背景等尚未见有研究报道。为此,本研究拟以mtDNA D-loop部分序列为分子标记,分析环翠峪猕猴群体的遗传多样性现状,探讨其可能的原始产地等,以期为该猕猴种群的科学管理和可持续利用提供可靠信息。
1 研究方法
1. 1 种群现状调查
1996年,河南荥阳市环翠峪风景名胜区开发者引入猕猴约40 只,以建立种群、开展生态旅游。2020年3—12月,采用访问调查法、直接计数法统计该猕猴群体的数量。
1. 2 样品采集、DNA 提取和序列扩增
采集14只环翠峪(Huancuiyu,HCY)地理单元猕猴个体的粪便样品。为避免同一个体的重复采样,首先对猕猴行为进行观察以锁定目标个体,继而基于粪便的大小、形状和颜色,并将间距lt; 2 m的粪便作为一个独立样品收集。然后,使用一次性灭菌手套,将新鲜粪便样品放入10 mL 的样品收集管中。记录采样点的海拔和经纬度等信息。最后,按照样品采集顺序编号,放入干冰保存。
粪便样品DNA的提取使用PowerFecal DNA Kit(QIAGEN, Germany),并参考已发表的mtDNA Dloop序列引物[33]:dloop-F(5′-TCCGAGGGCAATCAGAAAGAAA-3′)和dloop-R(5′-GCCTTGAGGTAAGAACCAGATGC-3′),进行PCR扩增。采用25. 0 μL反应体系,包括2. 0 μL 模板DNA、1. 5 μL 上游引物(10 μmol/L)、1. 5 μL下游引物(10 μmol/L)和20. 0 μL1. 1 × T3 Super PCR Mix(北京擎科生物科技股份有限公司)。反应条件:98 ℃预变性3 min;8 ℃变性10 s,55 ℃退火10 s,72 ℃延伸12 s,循环35次;最后72 ℃延伸5 min。将质量合格的PCR产物送至北京擎科生物科技股份有限公司测序。
1. 3 数据分析
结合本课题组已发表的、分布于河南济源王屋地区(Wangwu,WW)太行山猕猴的19 条mtDNA Dloop序列[34],以及NCBI 数据库中来自国内11 省份(河南、安徽、福建、浙江、湖北、湖南、贵州、广西、海南、四川和云南)的125条猕猴相关序列,进行后续分析。
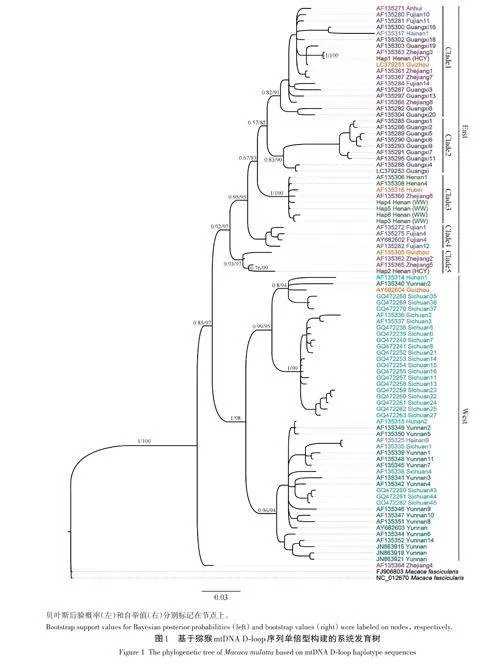
采用DNASTAR 软件包的SEQMAN 模块[35],查看并组装序列。为确保所测DNA序列为目的片段,将组装序列结果在NCBI数据库中与目标物种的同源序列进行比对。采用DnaSP v6. 12软件[36]统计变异位点(variable sites)、简约信息位点(parsimony informativesites)和单倍型数(haplotype number),计算群体的单倍型多样性(haplotype diversity,Hd)和核苷酸多样性(nucleotide diversity,π)。基于最大似然法(maximum likelihood,ML)和贝叶斯法(Bayesian inference,BI)构建系统发育树,以食蟹猴(Macaca fas⁃cicularis)为外群(GenBank登录号:FJ906803和NC_012670)。使用PhyloSuite v1. 2. 2[37]构建mtDNA Dloop单倍型的系统发育树。(1)序列比对:将“fasta”格式文件导入MAFFT进行序列比对。(2)ModelFinder:将比对完成的序列导入,“Criterion”选择Bayesian informationcriterion(BIC),“model for”选择MrBayes,其余均为默认参数,运行后可得到贝叶斯最佳模型。(3)构建贝叶斯树:运行设置为2 × 106代,每运行100次抽样一次,舍弃前25%的老化样本。(4)构建似然树:使用ModelFinder 选择最佳模型,BootStrap 选择Ultrafast,自举值设置为5 000。运行后使用FigTreev1. 4. 3[38]读取树结果文件,并进行可视化处理。使用PopART v1. 7软件[39]构建单倍型网络结构图。使用MEGA X软件[40]对序列进行比对,基于Kimura 2-parameter模型计算群体间遗传距离。使用Arlequinv3. 5软件[41]进行群体分子方差分析(analysis of molecularvariance,AMOVA),以估算遗传变异在不同地理单元猕猴群体间的分布。
2 结果
2. 1 种群数量
环翠峪猕猴群体自1996年引入,约40只。至调查结束时,计有4群约300只个体,其中约200只较为常见。
2. 2 序列特征和单倍型分布
环翠峪(HCY)地区14 只猕猴个体的D-loop 区序列(502 bp),存在25个单变异位点(5. 0%),且所有变异位点均为转换位点。D-loop序列碱基组成分析显示,A、T、C 和G 的平均含量分别为31. 1%、29. 8%、26. 5% 和12. 6%,其中A+T含量(60. 9%)明显高于G+C含量(39. 1%),表现出AT偏倚性。
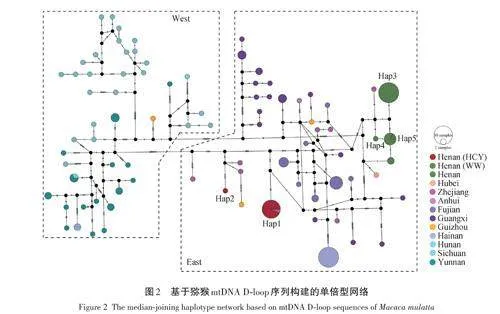
结合河南王屋(n = 19)以及NCBI 数据库(n =125)中猕猴D-loop序列,共定义86个单倍型(表1)。其中,Hap1在环翠峪和浙江猕猴群体间共享;湖南猕猴群体单倍型Hap35与云南猕猴群体共享;Hap38是海南和四川猕猴群体的共享单倍型。Hap2为环翠峪猕猴群体特有单倍型;河南、安徽、福建、广西、贵州和湖北猕猴群体间无共享单倍型。
2. 3 群体遗传多样性
各猕猴群体均表现出高的单倍型多样性[Hd: 0 ~(1. 000 ± 0. 500)]和低的核苷酸多样性[π: 0 ~(0. 065 20 ± 0. 019 36)](表2)。与其他地区相比,环翠峪猕猴群体核苷酸多样性水平(π: 0. 007 11 ±0. 005 92)明显高于河南猕猴群体,但低于除安徽和湖北之外的其他地理单元猕猴群体;另外,环翠峪地理单元猕猴群体单倍型多样性(Hd: 0. 143 ± 0. 119)低于上述猕猴群体,表明该猕猴群体单倍型类型较单一。
2. 4 系统发育和遗传结构
基于158条猕猴mtDNA D-loop序列所定义的86个单倍型,构建的ML 和BI 系统发育拓扑结构一致(图1)。系统发育分析结果表明,来自中国西部省份(云南和四川)和东部省份(河南、安徽、福建、浙江、湖北、湖南、贵州、广西和海南)的猕猴个体,分别聚为西部(West)和东部(East)遗传组(BI/PP = 0. 85/97)。东部遗传组又被划分为5个遗传支(Clade1~5),并得到较高的支持度。河南猕猴个体单倍型聚于Clade3支(BI/PP = 1/100)。环翠峪猕猴特有的单倍型Hap1和Hap2分别聚于Clade1和Clade5支内,且2个单倍型均与浙江猕猴个体表现出较近的亲缘关系(BI/PP = 1/100;BI/PP = 0. 76/99)。
单倍型网络分析结果显示,所有猕猴个体的86个单倍型可划分为2个单倍型簇,西部(West)和东部(East),并且2个单倍型簇间存在19个突变步数(图2)。环翠峪和河南猕猴群体均位于东部单倍型簇内,并得到系统发育结果的支持(图1)。其中,环翠峪猕猴群体单倍型Hap1与浙江猕猴群体共享;单倍型Hap2与浙江猕猴群体间虽然存在较多的突变步数和未知单倍型的遗传变异,但与其他猕猴群体相比,依然显示较近的亲缘关系。另外,环翠峪猕猴群体的2个特有单倍型(Hap1和Hap2)间也存在较高的突变步数,表明该猕猴群体的个体间存在较大的遗传差异(图2)。
2. 5 群体间遗传差异
群体间遗传距离分析表明,环翠峪猕猴群体与其他猕猴群体间的遗传距离为0. 034 8 ~ 0. 070 5(表3)。其中,环翠峪猕猴群体与浙江猕猴群体间的遗传距离最小(0. 034 8),而与海南猕猴群体存在较大的遗传差异(0. 070 5)。值得注意的是,环翠峪猕猴群体与河南猕猴群体间的遗传距离,均大于该猕猴群体与其他地区猕猴群体(海南群体除外)间的遗传距离(表3)。
环翠峪猕猴群体与东部组(河南、安徽、福建、广西、贵州、湖北、海南和浙江)内其他地区猕猴群体间的分子方差分析结果显示(表4),群体间和群体内的方差分别占总变异的63. 10%和36. 90%,表明遗传变异主要发生在群体间。
3 讨论
本研究基于猕猴158条mtDNA D-loop部分序列(502 bp)定义的86个单倍型,评估了河南荥阳环翠峪猕猴群体的遗传多样性水平,进而构建系统发育和单倍型网络。结果表明,环翠峪猕猴群体表现出低的核苷酸多样性和高的单倍型多样性;该猕猴群体与浙江猕猴的亲缘关系较近,与其他地区猕猴群体存在较大的遗传差异。
环翠峪猕猴群体存在低的遗传多样性水平,但明显高于河南猕猴群体遗传多样性。该现象或有两种解释:其一,具有多个遗传背景的猕猴群体遗传多样性水平明显高于野生猕猴群体[33];其二,不同遗传背景猕猴个体间的基因流共同促进了群体内遗传变异水平[34,42]。由于该猕猴群体建群时间短,且猕猴具有较长的世代间隔(10. 4 a)和较低的核苷酸突变率(0. 77 × 10-8)[43],故遗传漂变对该猕猴群体的遗传多样性水平影响较小。
环翠峪猕猴的初始种群可能来自浙江地区。猕猴自上新世期间横穿青藏高原东南缘横断山脉进入中国,并在更新世期间沿着东部和西部两条路线快速辐射到中国的大部分地区[44−45]。本研究发现,系统发育揭示中国猕猴可分为东部和西部两大遗传谱系,与基于分子标记[45]和基因组[46−47]的研究结果相一致。环翠峪猕猴群体聚于东部支,表明该猕猴群体源自中国东部地区。值得注意的是,环翠峪猕猴群体与其临近地理单元(如河南太行山地区、安徽和湖北)猕猴群体间存在较大的遗传差异,而与浙江猕猴群体存在较近的亲缘关系。
综上所述,环翠峪猕猴群体遗传多样性高于已知的国内其他野生猕猴群体,且为跨省份引入群体,为非自然分布,其原始种群分布区应为浙江地区。从中国猕猴的分类与分布来看[31],环翠峪猕猴与河南地区的太行山猕猴(猕猴华北亚种)分属于不同亚种。鉴于该猕猴群体发展迅速,一旦出现种群逃逸,将对华北地区野生太行山猕猴种群或有潜在的负面影响。因此,本研究建议,不应盲目引入动物用于生态旅游活动;在野生动物管理部门指导下,应对环翠峪猕猴群体采取严格的管控措施,限定其活动范围,以防溢出。
猜你喜欢
学苑创造·A版(2025年2期)2025-01-14 00:00:00
今日农业(2022年15期)2022-09-20 06:54:16
小猕猴学习画刊(2022年10期)2022-01-01 04:48:21
童话世界(2020年32期)2020-12-25 02:59:14
小猕猴智力画刊(2019年4期)2019-05-08 21:56:28
小猕猴智力画刊(2019年3期)2019-04-19 00:02:10
长江蔬菜·学术版(2016年12期)2017-01-12 20:57:59
中国科技博览(2016年25期)2016-12-20 20:04:30
农家科技下旬刊(2016年9期)2016-12-15 10:42:25
中国科技博览(2016年11期)2016-05-06 02:35:39
