
免疫性血栓性血小板减少性紫癜1 例报告及文献复习
2024-12-25 00:00:00王凌宇沈卫章谈磊李金梁
吉林大学学报(医学版) 2024年6期
[摘 要] 目的:分析1例免疫性血栓性血小板减少性紫癜 (iTTP) 患者的临床特点、干预时机、诊治方案和预后等因素,为该类罕见病的精准诊疗提供更多临床依据。方法:收集1例既往接受病毒灭活疫苗后早期被误诊为急性感染的iTTP 患者的临床资料,包括临床表现和辅助检查资料,并进行相关文献复习。结果:患者,男性,60岁,因“发热6 d”入院,查体下肢散在红色斑丘疹,早期仅有血小板轻度减少,入院时PLASMIC 评分为5 分。入院诊断急性感染,给予消炎、抗感染和激素等治疗效果不佳,1 周后复查血常规血小板明显减少,并随病情进展逐渐表现为严重的血小板减少、血尿、酱油色尿和神经及精神症状。完善相关检查,重新评估PLASMIC 评分为7 分。高度疑诊iTTP 后立即启动治疗性血浆置换(TPE),治疗过程中含血栓形成素1 型结构域的血管性血友病因子裂解酶ADAM 金属肽酶13 (ADATMS13) 活性水平lt;1%,ADAMTS13 抑制物检测结果为阳性,基因检测存在iTTP 易感基因的错义突变。诊断明确后规律应用利妥昔单抗静脉注射,完成4 个周期的治疗并随访至今,评估病情治疗有效。结论:iTTP常由于首诊临床症状不典型导致病情延误,一旦疑诊应立即启动以TPE 和糖皮质激素为基础的治疗,利妥昔单抗等新药为iTTP 多学科综合诊治策略提供了新选择。
[关键词] 血栓性血小板减少性紫癜; 临床分析; 血浆置换; 利妥昔单抗
[中图分类号] R736. 1 [文献标志码] B
血栓性血小板减少性紫癜(thromboticthrombocytopenic purpura,TTP) 是一种罕见而危重的血液系统疾病,通常发病率为每百万人中2~6 例[1]。TTP 的损伤机制是由于含血栓形成素结构域1 型基序的解整合素样金属蛋白酶13 (a disintegrinand metalloproteinase with thrombospondin type 1motif member 13, ADAMTS13) 的缺乏或功能障碍,导致血浆中未折叠的高相对分子质量血管性血友病因子(von Willebrand factor,vWF) 聚集,形成微血管血栓,引起多脏器缺血缺氧损害。TTP 分为2 种类型,由基因突变引起的血浆ADAMTS13活性严重缺乏称为遗传性或先天性TTP (congenitalTTP,cTTP);由抑制血浆ADAMTS13 活性的自身抗体免疫介导引发的TTP 称为免疫性TTP(immune TTP, iTTP), 约占TTP 的94. 5% [2],其特征是血小板低、贫血和微血管血栓形成。由于区域及医疗资源限制, 较多情况下无法立即获得ADAMTS13 活性、抑制物检测和基因检测等结果,首诊医生通常根据TTP 的典型临床表现和血液学检查结果进行诊断,患者典型临床表现包括微血管病性溶血、血小板减少症、神经系统症状、发热及肾脏损害等“五联征”,部分患者仅出现前3 项症状,称为“三联征”。由于上述临床症状不具有特异性,常常难以鉴别,导致临床诊疗病情延误而严重影响患者的预后和生存。国际上关于肺炎灭活病毒及疫苗相关性TTP 的研究较少, 且多与mRNA 疫苗相关, 国内尚未见病毒灭活疫苗相关报道[3]。本文作者报道1 例就诊于吉林大学第二医院既往接受病毒灭活疫苗后早期被误诊为急性感染的iTTP 患者的临床资料,并结合文献复习,分析其临床表现、实验室检查及影像学特点、理学特征和诊疗情况,探讨TTP 患者的临床病理特征及预后因素, 从而进一步提高临床医生对该疾病的认识。
1 临床资料
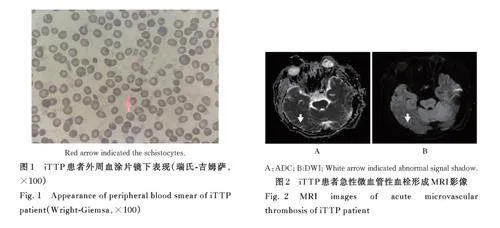
1. 1 一般资料 患者,男性,60岁,因“发热6 d”于2023 年2 月10 日入院,该患者入院6 d 前无明显诱因出现发热,体温最高达40 ℃,无咳嗽和咳痰,自行口服“扑热息痛”,体温略有下降。次日出现双下肢红色丘疹,伴发痒(患者自述对发痒丘疹抓挠,已至局部皮疹破溃和红肿),皮疹约黄豆大小,后逐渐向上蔓延至全身,就诊于本院急诊科,给予“依替米星、喜炎平和丙帕他莫”治疗后未见明显好转,后收入于本院急诊病房,体格检查双下肢散在红色丘疹。入院后提检血常规提示血红蛋白139 g·L-1 (115~150 g·L-1),血小板114×109 L-1(125~350×109 L-1), 平均红细胞体积82. 9 fL(82~100 fL), 乳酸脱氢酶512 U·L- 1 (120~250 U·L-1), 凝血酶原时间国际标准化比值(international normalized ratio, INR) 1. 14(0. 80~1. 20),C 反应蛋白261 mg·L-1(lt;5 mg·L-1), 降钙素原3. 04 μg·L-1 (0~0. 50 μg·L-1), 白细胞介素614. 72 ng·L-1 (0~6. 40 ng·L-1),间接胆红素、溶血筛查和尿常规检查未见明显异常。该患者于疾病早期被误诊为急性感染。随后继续行抗感染、消炎和对症支持等治疗,病情未见明显好转。1 周后复查血常规,提示血红蛋白及血小板进行性降低,血常规异常主要表现为血红蛋白65 g·L-1 (115~150 g·L-1),平均红细胞体积90 fL (82~100 fL),网织红细胞6. 46% (0. 61%~2. 20%), 血小板19×109 L-1 (125~350×109 L-1)。乳酸脱氢酶1 982 U·L-1 (120~250 U·L-1), 凝血酶原时间INR 1. 17 (0. 80~1. 20),出现血尿和轻微躁动。1. 2 实验室、影像学和其他检查 外周血涂片检查瑞氏-吉姆萨染色, 于100 倍镜下观察, 每张涂片计数200 个有核细胞, 可见数量不等的破碎红细胞。主要表现为不同程度的异型红细胞增多, 多由破碎红细胞形成, 可表现为泪滴状细胞和散在的裂片红细胞等。典型外周血涂片中破碎红细胞见图1。
因微血管栓塞所致的神经系统临床改变包括轻微性格改变和神经精神症状,患者行头部计算机断层扫描(computed tomography, CT) 检查, 提示多发点状和斑片模糊低密度影。行头部核磁共振成像(magnetic resonance imaging,MRI) 检查,可见新发的斑点状和片状的高信号, 表观扩散系数(apparent diffusion coefficient,ADC) 图呈低信号。见图2。上述表现与腔隙性脑梗塞难以鉴别,考虑与急性微血管性血栓形成有关, 但缺乏诊断的特异性。
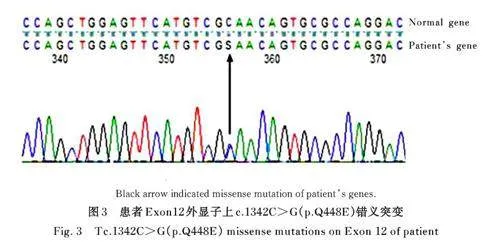
1. 3 诊断和治疗 疾病诊断参照 《血栓性血小板减少性紫癜诊断与治疗中国指南(2022 版) 》[4],其中PLASMIC 评分系统包括: 血小板计数减少、乳酸脱氢酶水平升高、贫血、中枢神经系统症状、多发性症状、血压、INR 和肌酐。每项指标符合以下标准给予1 分: ① 血小板计数lt;30×109 L-1;② 血清肌酐水平lt;176. 8 μmol·L-1; ③ 溶血证据(间接胆红素gt;34. 2 μ mol·L- 1 、网织红细胞计数gt;2. 5% 或结合珠蛋白消失);④无进展期癌症;⑤无实体器官或造血干细胞移植史; ⑥ 平均红细胞体积lt;90. 0 fL; ⑦ INRlt;1. 5。根据PLASMIC评分系统,总分0~4 分为低危,TTP 预测效率lt;5%; 总分5 分为中危,预测效率5%~25%;总分6~7 分为高危,预测效率60%~80%,该评分系统有助于在紧急情况下快速评估患者是否需要进一步地诊断和治疗,在及早诊治TTP 方面更具临床价值[5]。该患者入院时PLASMIC 评分为5 分,随后疾病进展,重新评估为7 分。2023 年2 月17 日高度疑诊为iTTP 后, 患者立即行治疗性血浆置换(therapeutic plasma exchange, TPE)。输入新鲜冰冻血浆2 000 mL, 每日2 次持续5 d, 同时联合甲泼尼龙80 mg静脉注射,每日2次,持续6 d。2023年2 月21 日辅助检验结果显示:ADAMTS13 活动度检测为1% (70%~120%, lt;10% 为重度减低),ADAMTS13 抑制物检测结果为阳性。ADAMTS13 基因检测结果显示: 考虑存在5 个变异, 其中4 个为同义变异, 该基因第12 外显子上c. 1342Cgt;G (p. Q448E) 为错义突变, 即基因遗传序列中第1 342 个核苷酸由胞嘧啶变异为鸟嘌呤,导致第448 个氨基酸由谷氨酰胺变成谷氨酸。具体基因改变见图3。后于2023 年2 月23 日开始规律应用利妥昔单抗静脉注射治疗,共计4 个周期,疗效评估参照国际TTP 工作组修订iTTP 治疗结局定义[6]。完成治疗后,患者复查血象恢复和临床症状消失, 随访至2023 年12 月患者复查无明显异常,临床治疗有效。
2 讨 论
TTP 病因和病情复杂, 可由感染、药物、免疫功能紊乱、妊娠、肿瘤和骨髓移植等多种原因诱发。TTP 由于发病率较低, 疾病发展迅速, 临床和辅助检查等表现不具有特异性。在实际工作中,患者通常未及时行ADAMTS13 活动度及抑制物检测,或因其检测结果呈现较慢,加之临床医生认识不足,往往导致误诊和漏诊率较高。因此,首诊医生必须熟知TTP 的临床表现,并在紧急情况下做出迅速和准确的判断,从而正确干预及指导治疗。除“ 五联征” 和“ 三联征” 外, TTP 首诊时其他非特异性表现包括乏力、腹痛、恶心甚至呕吐等,增加了鉴别诊断难度。其中,中度发热在诊断时并不常见,但神经系统体征是诊断的基本要素,60%的患者常表现为伴有头痛、抽搐和短暂性脑缺血发作,如轻偏瘫、失语症、构音障碍和黑矇,也可能伴有意识模糊及昏迷等情况。由于以上神经症状常为一过性表现,易被患者和临床医生忽视,因此更应该详细及系统地询问病史。肾脏受累在该疾病中并不常见,血清肌酐中度升高且低于200 μmol·L-1、血小板减少且低于20×109 L-1 被认为是预后不良因素[7]。贫血常表现为正细胞正色素性的溶血性贫血, 伴间接胆红素、乳酸脱氢酶水平升高和结合珠蛋白表达减少,Coombs 试验阴性。诊断的关键要素是在有核细胞减少的情况下, 在血涂片检查中发现破碎红细胞。然而,破碎红细胞可能在病程最初24~48 h 内消失,因此有必要在疾病早期行骨髓穿刺和外周血涂片, 从而避免相关支持证据的遗漏[8]。
对于iTTP 或cTTP 患者, 因其早期临床症状不典型而难以区分,临床上通常采用ADAMTS13活性和自身抗体检测。当患者抗ADAMTS13 自身抗体检测呈阳性时,可诊断为iTTP。当ADAMTS13活性明显降低至lt;10% 且抗ADAMTS13 自身抗体呈阴性时, 怀疑cTTP。一旦疑诊cTTP, 应在患者父母和子女中评估ADAMTS13 活性, 并进行ADAMTS13 基因分析,以进行明确诊断[9]。该患者于发病前1 个月应用肺炎病毒灭活疫苗后出现皮疹及发热,并在病程的1 周后出现初次TTP 症状。有关肺炎感染及疫苗相关报道较少, 但有研究[10]显示:疫苗本身或其佐剂成分会产生自身抗体,可诱导免疫系统异常应答, 从而导致自身免疫性疾病。该患者不除外肺炎灭活病毒疫苗接种后加速TTP 发生发展的可能。该患者存在ADAMTS13 单一核苷酸多态性突变,此修饰性变异虽不明显改变ADAMTS13 活性, 但可调控其他病理性变异的活性[11]。同时检测到患者女儿Exon21 外显子上c. 2708Cgt;T (p. S903L) 的错义突变, 虽未在首次注射肺炎病毒灭活疫苗后发病, 但不除外该患者TTP 的遗传易感性增加而在再次接种灭活病毒疫苗后诱发致病可能。因此在接种肺炎灭活病毒疫苗前有必要全面了解既往史及家族史,尤其对于存在自身免疫性疾病、肿瘤、感染和免疫抑制状态等情况的患者,还应考虑接种疫苗后的及时监测和临床随访。
TPE 是目前最常用和首选的治疗TTP 的方法,延迟开始TPE 会使预后恶化,TPE 不仅能够补充血浆中ADAMTS13 活性, 清除抗ADAMTS13 自身抗体, 还可清除异常的超大相对分子质量的vWF 多聚体[12-13]。研究[14]显示:在疾病确诊后8 h 内和24 h 内行TPE, 患者死亡风险无明显差异, 而延迟gt;24 h 行TPE,患者死亡率和主要血栓事件明显增加。研究[15-16] 显示: TPE 治疗次数对患者预后无明显影响,可以根据患者的临床表现及实验室检查结果进行选择,症状及实验室检查结果持续不缓解者给予更多次数的TPE。但TPE 常因地区、技术限制和经济条件限制无法及时应用。研究[17]显示:新鲜冰冻血浆(fresh frozen plasma, FFP)、用溶剂或洗涤剂病毒减毒的血浆(solvent/detergent fresh frozen plasma, SD-FFP) 和冷冻上清液血浆(cryosupernatant plasma,CP) 均含有等效的ADAMTS13 活性。而FFP 可作为普遍接受的预防和治疗早期TTP 的方法之,常用剂量为15 mL·kg-1,每3~5 d 或每周1 次[18]。
iTTP 常为原发疾病逐渐进展后出现的首发表现。30% iTTP 患者的演变特征为复发和缓解期交替, 称为慢性复发或复发性TTP[19]。研究[20] 显示: 20%~50% 的iTTP 患者在发病后的第1 年甚至更晚出现急性期缓解后的复发。在经过5 次TPE 联合糖皮质激素治疗无临床反应的患者称为难治性TTP[4]。复发性和难治性TTP 患者对TPE的标准治疗反应缓慢且不完全。尽管TPE 和免疫抑制剂及免疫调节疗法改善了患者的预后, 但TTP 的死亡率仍然较高, 难治性患者的死亡率甚至更高[21]。
目前多种新型辅助疗法提供了TTP 新的诊疗思路。利妥昔单抗作为一种靶向 B 淋巴细胞CD20 抗原的单克隆抗体,目前用于治疗B 淋巴细胞肿瘤形成和自身免疫性疾病。研究[22] 显示:利妥昔单抗通过清除循环中的B 淋巴细胞和减少抗ADAMTS13 自身抗体的产生而发挥作用,可降低iTTP 复发率,延长复发间期和发作间隔期。单药治疗可使大量难治和复发性TTP 患者获得缓解。既往应用的脾切除和高剂量免疫球蛋白已逐渐被利妥昔单抗所替代[23]。目前常用治疗策略包括加强至每日2 次TPE 联合使用利妥昔单抗或其他免疫抑制。而当利妥昔单抗与TPE 联合使用时, 需要考虑到发病早期肾脏微血管血栓形成影响清除率,理想情况下可在TPE 后给药。
此 外,治疗后定期监测血小板数目、ADAMTS13 活性和自身抗体有助于更好地随访iTTP 患者是否进展为复发和难治性TTP[24]。研究[25] 发现:TTP 患者认知异常和抑郁的频率明显高于正常人群,可伴有卒中和神经认知障碍,如持续记忆和注意力问题、轻微认知异常和抑郁等长期不良反应。因此, 在iTTP 患者急性发作恢复后,有必要长期监测其认知功能和抑郁状态。该患者应用TPE 联合利妥昔单抗4 周期治疗后症状完全缓解,随访至2023 年12 月无复发相关迹象及神经认知障碍,但仍需长程随访及监测。然而对于在利妥昔单抗应用以外,管理难治性或复发性iTTP 的方法和干预时机尚无循证或共识指南。最佳给药剂量和给药间隔等仍需扩大样本进行更多前瞻性研究加以证实。
其他新型药物主要包括奥法木单抗和奥妥珠单抗,是完全人源化的第二代抗CD20 抗体,对因药物超敏反应等不良反应而对利妥昔单抗不耐受的iTTP 患者有效且安全[26]。抗免疫系统药物卡普赛珠单抗作为一种人源化的单可变域免疫球蛋白片段,特异性靶向结合VWF 的A1 结构域,阻断其与血小板糖蛋白GPⅠb-Ⅸ-Ⅴ复合物(glycoproteinⅠ b-Ⅸ -Ⅴ complex, GP1b-Ⅸ -Ⅴ) 受体的相互作用[27]。这种vWF 纳米抗体可抑制血小板与vWF 结合并防止微血栓形成。已被证实可与标准治疗相结合改善iTTP 的预后, 为iTTP 提供了临床治疗的新选择[27]。然而vWF 纳米抗体药物价格昂贵,可获率相对较低[28]。此外,对于急性发作期iTTP 患者,尽管目前其总生存率有所提高,但基于TPE 的治疗仍存在多种风险,包括过敏反应、导管相关并发症和容量超负荷,尤其是血浆输注等。而新型重组人ADAMTS13 (recombinant human ADAMTS13,rhADAMTS13) 靶向治疗可使严重的TTP 症状减轻, 并可能使治疗iTTP 所需的TPE 次数频率减少,rhADAMTS13 的靶向治疗可在无需依赖TPE条件下尝试针对TTP 的根本病因治疗,对于iTTP未来治疗策略具有一定的价值[29]。
该患者初治时误诊,分析可能原因如下:以并发急性感染的血小板减少或原发病为首发表现的患者就诊于非血液病科室,大部分患者因非特异性表现导致诊疗延迟,在疾病治疗过程中部分患者自动放弃治疗或出现重症感染和脑出血等并发症,导致患者预后差,甚至死亡。当患者表现为微血管病性溶血性贫血并发血小板减少,且在ADAMTS13 活性无法及时获得时,可根据PLASMIC 评分系统协助诊断TTP,从而尽早实施治疗。早期应用TPE和利妥昔单抗是改善预后的关键。同时随着新药问世, 以TPE、糖皮质激素、利妥昔单抗和卡普赛珠单抗组成的“ 四联” 疗法,已被许多国家纳入iTTP 的治疗标准[30]。目前,我国的临床数据有待进一步更新,为iTTP 多学科及综合诊治策略提供更多的选择和方案。
利益冲突声明:所有作者声明不存在利益冲突。
作者贡献声明:王凌宇和沈卫章参与研究设计及论文撰写,谈磊参与数据收集和分析及论文撰写,李金梁参与研究设计、论文撰写和审校。
[参考文献]
[1] STALEY E M, CAO W J, PHAM H P, et al. Clinical
factors and biomarkers predict outcome in patients with
immune-mediated thrombotic thrombocytopenic
purpura[J]. Haematologica, 2019, 104(1): 166-175.
[2] JOLY B S, COPPO P, VEYRADIER A. Thrombotic
thrombocytopenic purpura[J]. Blood, 2017, 129(21):
2836-2846.
[3] SALUJA P, GAUTAM N, YADALA S, et al.
Thrombotic thrombocytopenic purpura (TTP) after
COVID-19 vaccination: a systematic review of reported
cases[J]. Thromb Res, 2022, 214: 115-121.
[4] 中华医学会血液学分会血栓与止血学组. 血栓性血小板
减少性紫癜诊断与治疗中国指南(2022年版)[J]. 中华
血液学杂志, 2022, 43(1): 7-12.
[5] LI A, KHALIGHI P R, WU Q, et al. External
validation of the PLASMIC score: a clinical prediction
tool for thrombotic thrombocytopenic purpura diagnosis
and treatment[J]. J Thromb Haemost, 2018, 16(1):
164-169.
[6] CUKER A, CATALAND S R, COPPO P, et al.
Redefining outcomes in immune TTP: an international
working group consensus report[J]. Blood, 2021,
137(14): 1855-1861.
[7] ROCK G, KELTON J G, SHUMAK K H, et al.
Laboratory abnormalities in thrombotic
thrombocytopenic purpura. Canadian Apheresis
Group[J]. Br J Haematol, 1998, 103(4): 1031-1036.
[8] ALLFORD S L, HUNT B J, ROSE P, et al.
Guidelines on the diagnosis and management of the
thrombotic microangiopathic haemolytic anaemias[J].
Br J Haematol, 2003, 120(4): 556-573.
[9] MATSUMOTO M, FUJIMURA Y, WADA H, et al.
Diagnostic and treatment guidelines for thrombotic
thrombocytopenic purpura (TTP) 2017 in Japan[J].
Int J Hematol, 2017, 106(1): 3-15.
[10]GIUFFRIDA G, CONDORELLI A, DI GIORGIO M A,
et al. Immune-mediated thrombotic thrombocytopenic
purpura following administration of Pfizer-BioNTech
COVID-19 vaccine[J]. Haematologica, 2022, 107(4):
1008-1010.
[11]STOLL M, RÜHLE F, WITTEN A, et al. Rare
variants in the ADAMTS13 von willebrand factorbinding
domain contribute to pediatric stroke[J]. Circ
Cardiovasc Genet, 2016, 9(4): 357-367.
[12]PEREIRA A, MAZZARA R, MONTEAGUDO J, et al.
Thrombotic thrombocytopenic purpura/hemolytic uremic
syndrome: a multivariate analysis of factors predicting
the response to plasma exchange[J]. Ann Hematol,
1995, 70(6): 319-323.
[13]MATSUMOTO M, YAGI H, ISHIZASHI H, et al.
The Japanese experience with thrombotic
thrombocytopenic purpura-hemolytic uremic
syndrome[J]. Semin Hematol, 2004, 41(1): 68-74.
[14]SAWLER D, PARKER A, BRITTO J, et al. Time
from suspected thrombotic thrombocytopenic purpura to
initiation of plasma exchange and impact on survival:
a 10-year provincial retrospective cohort study [J].
Thromb Res, 2020, 193: 53-59.
[15]JOLY B S, COPPO P, VEYRADIER A. An update on
pathogenesis and diagnosis of thrombotic
thrombocytopenic purpura[J]. Expert Rev Hematol,
2019, 12(6): 383-395.
[16]SCULLY M, HUNT B J, BENJAMIN S, et al.
Guidelines on the diagnosis and management of
thrombotic thrombocytopenic purpura and other
thrombotic microangiopathies[J]. Br J Haematol, 2012,
158(3): 323-335.
[17]SADLER J E. Pathophysiology of thrombotic
thrombocytopenic purpura[J]. Blood, 2017, 130(10):
1181-1188.
[18]ASMIS L M, SERRA A, KRAFFT A, et al.
Recombinant ADAMTS13 for hereditary thrombotic
thrombocytopenic purpura[J]. N Engl J Med, 2022,
387(25): 2356-2361.
[19]SADLER J E, MOAKE J L, MIYATA T, et al.
Recent advances in thrombotic thrombocytopenic
purpura [J]. Hematology Am Soc Hematol Educ
Program, 2004: 407-423.
[20]KREMER HOVINGA J A, VESELY S K,
TERRELL D R, et al. Survival and relapse in patients
with thrombotic thrombocytopenic purpura[J]. Blood,
2010, 115(8): 1500-1511;quiz1662.
[21]VERBEKE L, DELFORGE M, DIERICKX D.
Current insight into thrombotic thrombocytopenic
purpura[J]. Blood Coagul Fibrinolysis, 2010, 21(1):
3-10.
[22]SAYANI F A, ABRAMS C S. How I treat refractory
thrombotic thrombocytopenic purpura[J]. Blood, 2015,
125(25): 3860-3867.
[23]BLOMBERY P, SCULLY M. Management of
thrombotic thrombocytopenic purpura: current
perspectives[J]. J Blood Med, 2014, 5: 15-23.
[24]SCHLEINITZ N, EBBO M, MAZODIER K, et al.
Rituximab as preventive therapy of a clinical relapse in
TTP with ADAMTS13 inhibitor[J]. Am J Hematol,
2007, 82(5): 417-418.
[25]DEFORD C C, REESE J A, SCHWARTZ L H, et al.
Multiple major morbidities and increased mortality during
long-term follow-up after recovery from thrombotic
thrombocytopenic purpura[J]. Blood, 2013, 122(12):
2023-2029;quiz2142.
[26]ROBERTZ J, ANDRES M, MANSOURI
TALEGHANI B, et al. Obinutuzumab in two patients
suffering from immune-mediated thrombotic
thrombocytopenic purpura intolerant to rituximab[J].
Am J Hematol, 2019, 94(10): E259-E261.
[27]CALLEWAERT F, ROODT J, ULRICHTS H, et al.
Evaluation of efficacy and safety of the anti-VWF
Nanobody ALX-0681 in a preclinical baboon model of
acquired thrombotic thrombocytopenic purpura [J].
Blood, 2012, 120(17): 3603-3610.
[28]TSE B, BUCHHOLZ M, PAVENSKI K.
Management of immune thrombotic thrombocytopenic
purpura with caplacizumab: a Canadian, single-centre,
real-world experience[J]. Platelets, 2023, 34(1):
2157807.
[29]MORONITI J J, VRBENSKY J R, NAZY I, et al.
Targeted ADAMTS-13 replacement therapy for
thrombotic thrombocytopenic purpura [J]. J Thromb
Haemost, 2024, 22(4): 896-904.
[30]ZHENG L, ZHENG X L. How should caplacizumab be
used for treatment of immune thrombotic
thrombocytopenic purpura?[J]. Ann Blood, 2023,
8: 11.
猜你喜欢
中国实用医药(2016年23期)2016-12-26 09:49:32
糖尿病新世界(2016年16期)2016-12-09 02:43:36
中国实用医药(2016年28期)2016-12-07 08:22:16
中国实用医药(2016年27期)2016-11-30 12:25:34
中国医药导报(2016年25期)2016-11-30 07:43:53
中外医学研究(2016年26期)2016-11-30 03:06:27
中外医学研究(2016年24期)2016-11-30 01:53:21
医学信息(2016年30期)2016-11-28 20:48:33
中外医学研究(2016年28期)2016-11-28 12:41:55
医学信息(2016年29期)2016-11-28 10:12:43
