
基于简化基因组开发青冈和滇青冈微卫星引物
2024-12-12 00:00:00欧阳泽怡李志辉牟虹霖姜小龙程勇吴际友
南京林业大学学报(自然科学版) 2024年6期

摘要:【目的】青冈(Quercus" glauca)和滇青冈(Q. glaucoides)为珍贵树种,是东亚亚热带常绿阔叶林的重要建群种以及典型地理替代种,具有很高的生态和经济价值。开发其微卫星(simple sequence repeats,SSR)引物有助于分析其群体遗传格局及遗传多样性,为2种青冈林的管理和资源开发利用提供参考,也可为跨物种间的微卫星标记开发提供借鉴。【方法】分别基于3株青冈和3株滇青冈植株的简化基因组测序数据开发SSR引物。先后采用Stacks 2.0b软件process_radtags模型和SciRoKo 3.4软件对测序数据进行过滤和提取。利用Primer premier 6.0软件设计SSR引物。【结果】使用pyRAD 3.0.66软件对序列进行聚类,共鉴定出217个SSR位点,其中35%(76个)的SSR位点在青冈和滇青冈中均具有多态性。设计并筛选出28对SSR引物,分别在两个青冈群体和两个滇青冈群体(共48个个体)中进行巢式聚合酶链式反应(PCR)扩增。开发的28对SSR引物分布在青冈10条染色体上,SSR引物均在青冈和滇青冈个体中成功扩增,扩增率达到了90.7%。SSR分型分析结果表明,从遗传多样性方面看,所开发的28对引物中,共有176个等位基因;引物的等位基因数为3~13,平均为6.29个;期望杂合度为0.223~0.886,观察杂合度为 0.159~0.830。【结论】利用简化基因组数据可以快速、高效、经济地开发青冈和滇青冈通用微卫星引物,为后续2种青冈群体遗传学研究提供基础。也证明利用简化基因组测序数据,可开发近缘物种的通用微卫星引物。
关键词:青冈; 滇青冈; 微卫星; 简化基因组
中图分类号:S795"""""" 文献标志码:A开放科学(资源服务)标识码(OSID):
文章编号:1000-2006(2024)06-0062-09
Designing of microsatellite primers for" Quercus glauca" and"" Q. glaucoides (Fagaceae) based on" RAD-seq data
OUYANG Zeyi1,2, LI Zhihui1, MOU Honglin1, JIANG Xiaolong1, CHENG Yong3, WU Jiyou3*
(1.College of Forestry, Central South University of Forestry and Technology, Changsha 410116," China;2. Hunan Botanical Garden, Changsha 410116, China; 3. Hunan Academy of Forestry, Changsha 410004," China)
Abstract:
【Objective】Quercus glauca" and Q. glaucoides are valuable and dominant species in the subtropical evergreen broadleaf forests of East Asia. They represent typical geographical vicarious species with significant ecological and economic importance. Therefore, the development of SSR primers for these two species can facilitate the analysis of genetic patterns and genetic diversity for the management and resource development of evergreen broadleaf forests as well as provide a reference for the development of microsatellite markers across species. 【Method】This study developed SSR primers based on RADseq data from three Q. glauca and three Q. glaucoides individuals, respectively. The sequencing data were filtered and extracted using the process_radtags model in Stacks 2.0b software and SciRoKo 3.4 software sequentially. SSR primers were designed using Primer premier V6.0 software.【Result】The sequences were clustered using pyRAD 3.0.66, identifying a total of 217 SSR loci, 35% (76) of which were polymorphic in both species. Twenty-eight SSR primer pairs were designed"" and validated in two Q. glauca populations and two Q. glaucoides populations (48 individuals in total) through nested polymerase chain reaction (PCR) amplification. The 28 SSR primer pairs are distributed across ten chromosomes of the Q. glauca genome and successfully amplified in Q. glauca and Q. glaucoides individuals, with an amplification rate of 90.7%. The SSR genotyping analysis detected a total of 176 alleles, with the number of alleles per primer ranging from 3 to 13, and an average of 6.29. The expected and observed heterozygosity of the primers ranged from 0.223 to 0.886 and from 0.159 to 0.830, respectively. 【Conclusion】The universal microsatellite primers developed in this study using RADseq data for Q. glauca and Q. glaucoides provide a basis for further population genetics studies of these species. In addition, this study demonstrates that RADseq data can be employed to rapidly, efficiently, and be used to" cost-effectively develop universal microsatellite primers for closely related species.
Keywords:Quercus glauca; Quercus glaucoides; microsatellite; RAD-seq
栎属(Quercus)约有450种,在北半球不同生境中广泛分布,是温带落叶阔叶林及热带、亚热带常绿阔叶林的优势树种[1]。国家林业和草原局的森林调查数据显示,栎类植物的分布面积和生物量大,占中国森林的10%以上。栎属植物在木材供应、环境绿化、涵养水源和水土保持等方面起到十分重要的作用[2]。因此,研究栎属的分类、种群结构及遗传历史也成为诸多学者关注的热点[3]。根据最新的分类系统,栎属主要包括栎亚属(Subgen. Quercus)和栓皮栎亚属 (Subgen. Cerris)。栓皮栎亚属包括3个组:Sect. Cyclobalanopsis、Cerris和Ilex[1]。青冈组(Sect. Cyclobalanopsis)是栓皮栎亚属的基部类群,约有100种,中国西南地区是青冈组的物种多样性中心[4]。青冈(Quercus glauca)和滇青冈(Q. glaucoides)隶属于栎属青冈组(Q. sect. Cyclobalanopsis),是东亚亚热带常绿阔叶林重要建群种,也是中国-喜马拉雅植物亚区和中国-日本森林亚区间的典型地理替代种[4]。滇青冈分布的海拔普遍比青冈高,在滇中高原南缘还存在小范围的镶嵌分布[5]。两者在形态上极为相似,但存在有少量稳定的分类特征[4]:如青冈的叶多为倒卵形、叶被为平伏“丁字毛”、壳斗外壁毛被较为稀疏;滇青冈叶为卵形,叶被的“丁字毛”长而卷曲,壳斗外壁具有明显的灰黄色毛。化石数据显示,这2种自中新世以来就广布于东亚亚热带[6-7]。这2种都具有重要的经济和生态价值,了解、比较其遗传多样性、遗传结构及遗传分化对青冈和滇青冈种质资源的开发与利用具有重要指导意义。
微卫星(microsatellite,MS)又称简单序列重复(simple sequnce repeat,SSR),广泛分布于真核生物和部分原核生物的基因组中[8]。SSR遗传标记自20世纪90年代始,就应用于鉴定亲子关系、亲缘关系以及种质资源[9]。利用高通量测序数据是目前开发SSR引物的主要方法。简化基因组测序数据为基因组信息缺乏物种的SSR引物开发提供了可能性[10]。此外,简化基因组测序技术可直接利用测序序列设计SSR引物;仅需通过1次测序即可获得数以万计的多态性标记,得到更多的SSR位点[11]。因此,基于简化基因组测序数据开发SSR引物的方法被应用于高等植物的SSR引物开发中,例如岭南青冈(Q. championii)[12]、金银花(Lonicera japonica)[13]、疣柄魔芋(Amorphophallus paeoniifolius)[14]、花锚(Halenia corniculata)[15]等植物。不过,目前的高通量测序开发SSR引物主要针对单个物种,对开发适用于不同物种的SSR通用引物的研究还较少。
本研究旨在基于简化基因组测序数据,开发适用于青冈和滇青冈群体的SSR引物。青冈和滇青冈作为中国亚热带常绿阔叶林建群种,开发群体的通用SSR引物可以用于其群体遗传格局及遗传多样性的探究,进而对常绿阔叶林的管理及经营提供参考。此外,进一步研究对揭示中国-日本植物亚区和中国喜马拉雅植物亚区的物种形成与演化也具有重要意义。同时,该研究也可为跨物种的SSR引物开发提供参考。
1 材料与方法
1. 1 建库以及实验验证样本
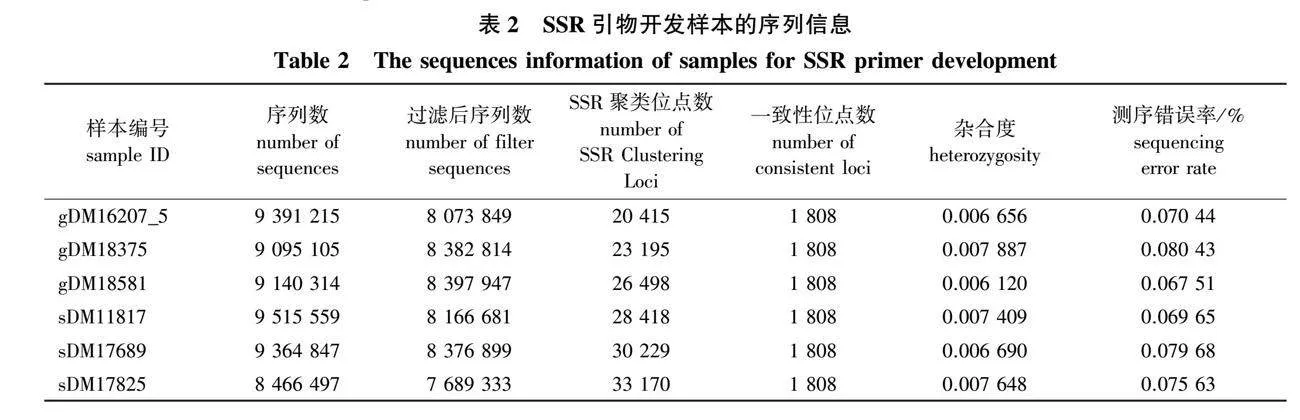
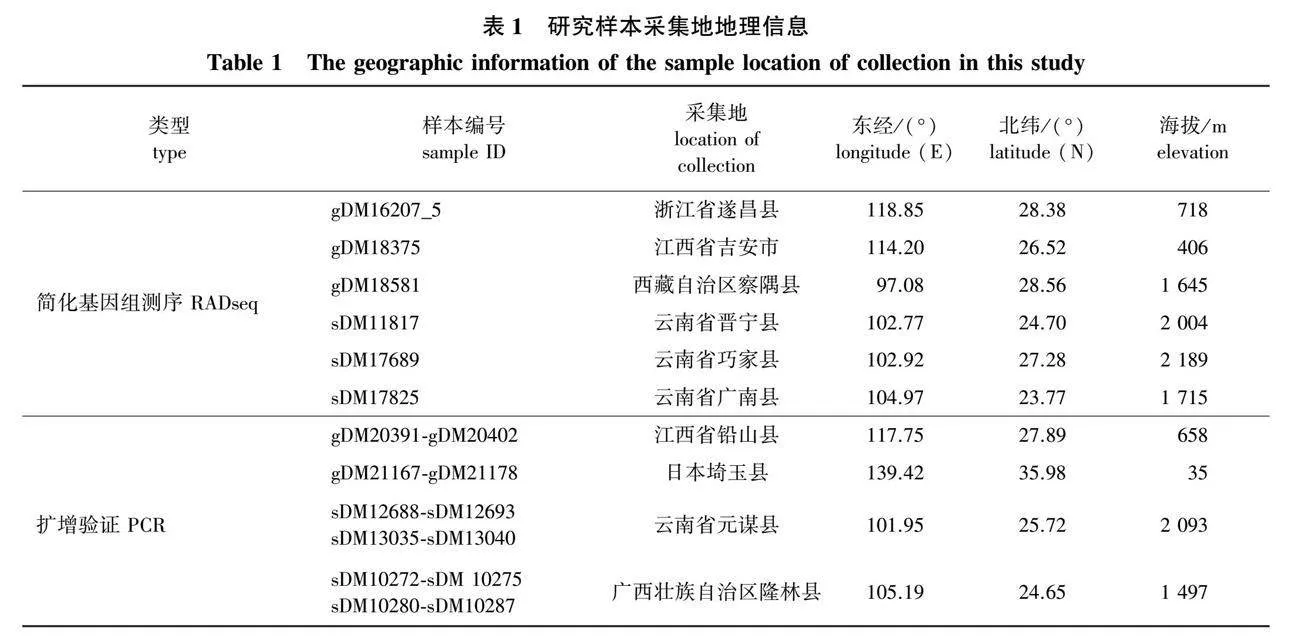
选择青冈和滇青冈各3株样本进行SSR引物设计。为获得更多的遗传变异信息,6株样本来自不同群体。滇青冈3株样本分别来自云南省晋宁区、巧家县和广南县群体;青冈3株样本来自浙江省遂昌县、江西省吉安市和西藏自治区察隅县群体。另选择青冈和滇青冈2个群体,每群体12单株对设计的SSR引物进行验证。滇青冈群体来自广西壮族自治区隆林县和云南省元谋县;青冈群体采自于江西省铅山县和日本埼玉县。样本采集地地理信息见表1。
1.2 简化基因组测序数据
使用DNeasy植物组织试剂盒(天根生化科技有限公司,北京)对二氧化硅干燥后的样本叶片进行总DNA提取。利用限制性内切酶Taq I(5′-T | CGA-3′)和Mse I(5′-T | TAA-3′)对提纯后的总DNA进行酶切,构建双酶切测序基因分型(ddGBS)文库。利用Illumina NovaSeq 6000基因测序仪(美国)对长度为400~600 bp的片段进行测序,测序模式为双端150 bp(PE150)。
1.3 通用引物设计
为获得高质量测序数据,采用Stacks 2.0b软件process_radtags模型对原始Reads进行过滤[16]。采用SciRoKo 3.4软件[17]提取包含SSR序列的Reads。采用pyRAD 3.0.66软件[18]对提取的reads进行聚类,个体内部和个体之间聚类时的碱基相似性阈值设为70%,每个聚类的最大插入缺失碱基数设为30。使用覆盖深度大于3个序列的聚类进行进一步分析。为了避免某些个体的PCR扩增失败,去除SSR侧翼序列中的转换/颠换变异等位点。为确保侧翼序列中有足够的碱基用于引物设计,仅保留SSR重复区位于26~129 bp、重复区等位基因数≥2的位点用于引物设计。利用Primer premier 6.0[19]软件设计SSR引物,设计好的引物由生工生物工程(上海)股份有限公司合成。为了获得设计引物在染色体上的分布,使用BLAST将设计好的引物序列比对至青冈基因组上。
1.4 实验与分析验证
对待实验的正向引物5′ 端添加通用接头序列M13(序列为TGTAAAACGACGGCCAGT)[20]。以提取的总DNA为模板,使用带 M13 接头的引物进行PCR扩增,利用添加带荧光标记的M13引物对第1轮扩增产物进行2次扩增。
PCR扩增体系如下:
第1轮PCR扩增体系为20 μL:2×Power Taq PCR MasterMix 10 μL、正向F引物(10 μmol/L) 1 μL、反向R引物(10 μmol/L) 1 μL、植物样本DNA 模板2 μL、ddH2O 6 μL。
以第1轮PCR产物为模板,继续第2轮PCR扩增,扩增体系为2 μL:Taq 酶(5 U/μL) 0.4 μL、10×PCR buffer 0.1 μL、带荧光标记的M13引物(10 ng/μL) 0.5 μL、ddH2O 1.0 μL。
PCR反应程序为94 ℃预变性 5 min;94 ℃变性 45 s, 57 ℃(第2轮PCR反应的退火温度为57.1 ℃)退火45 s, 72 ℃延伸45 s,35个循环;72 ℃延伸10 min,16 ℃保存。
PCR扩增结果经琼脂糖凝胶电泳检测合格后,由生物公司使用自动测序仪ABI 3730(美国,应用生物系统公司)测定产物长度。每个SSR标记的等位基因长度使用Genemarker 2.2.0软件进行基因分型[21]。分别由GenAlEx 6[22]和Cernicalin 1.30[23]计算每对引物和4个群体的遗传多样性,包括等位基因数(NA)、期望杂合度(He)和观察杂合度(Ho)。
2 结果与分析
2.1 基于简化基因组的青冈和滇青冈SSR位点开发
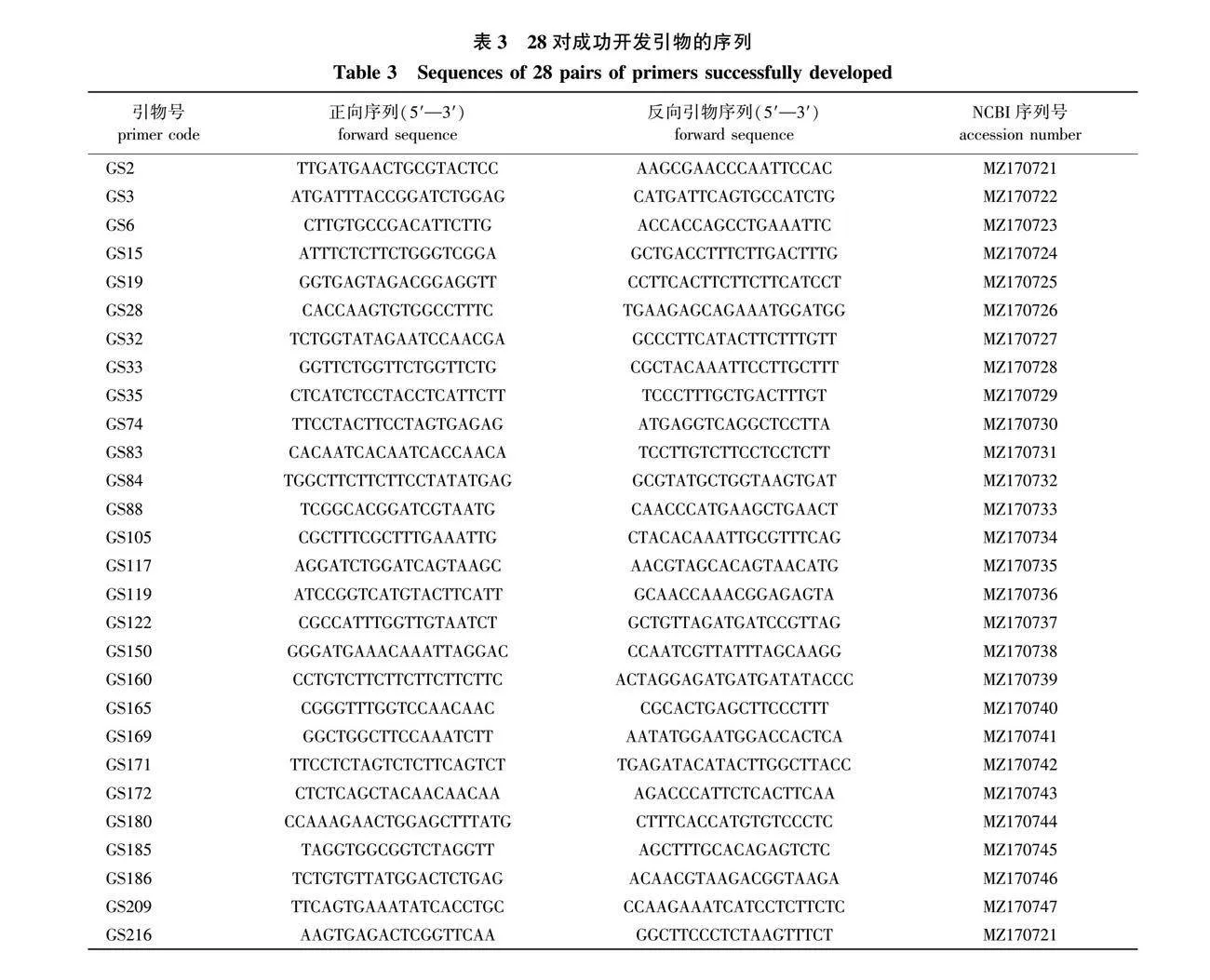
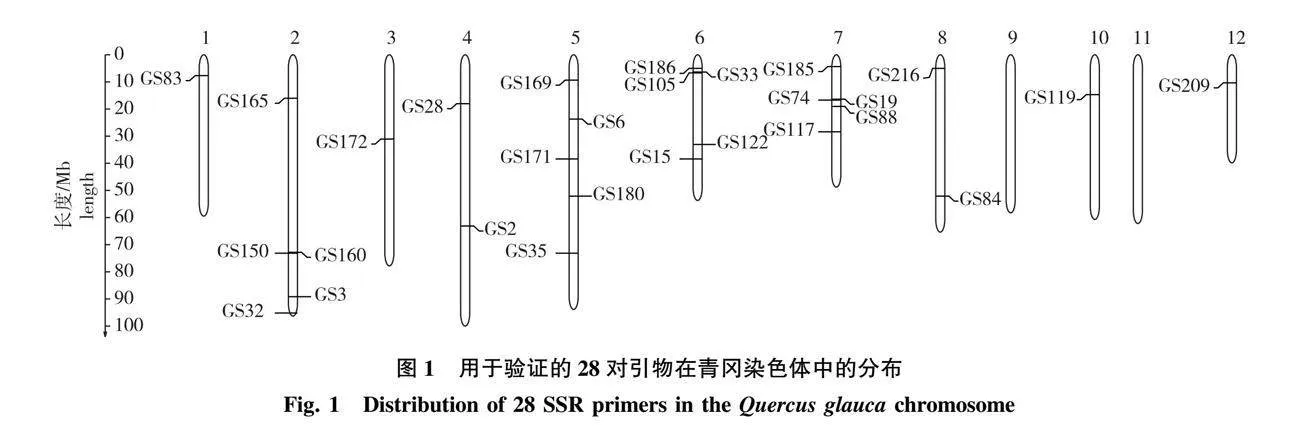
6个选定样本的原始测序序列范围为850万~940万条,过滤低质量数据后,序列范围为770万~840万条。提取出含有SSR位点的序列,对样本进行聚类,并筛选出同源序列后,每个样本中的基因座数量为20 415~33 170。在对所有样本进行样本间聚类,结果共获得1 808个位点(一致位点,表2)。使用Python脚本,对转换/颠换序列进行过滤,读取位于前25或129 bp的SSR重复序列以及单碱基重复SSR,最终获得217个位点。利用Primer premier 6.0设计了58对引物,筛选出28对引物(表3)进行实验验证。这28对引物位于青冈基因组12条染色体中的10条上,其中2号、5号、6号和7号染色体包含SSR位点最多(均5个),9号和11号没有SSR位点分布(图1)。
2.2 青冈和滇青冈SSR通用引物分析
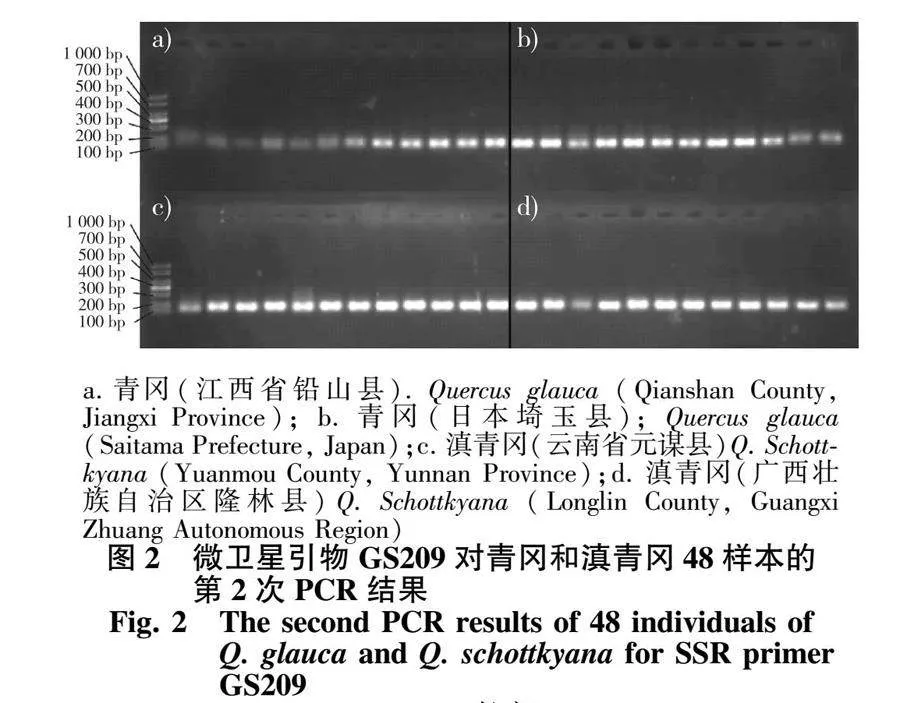
利用设计的28对通用引物对4个群体的48个样本进行PCR扩增。在两轮PCR扩增后进行琼脂糖凝胶电泳检测(以引物GS209为例,Marker均为1 000 bp,如图2所示),检测合格的PCR产物送至生工生物工程公司进行毛细管电泳分型。
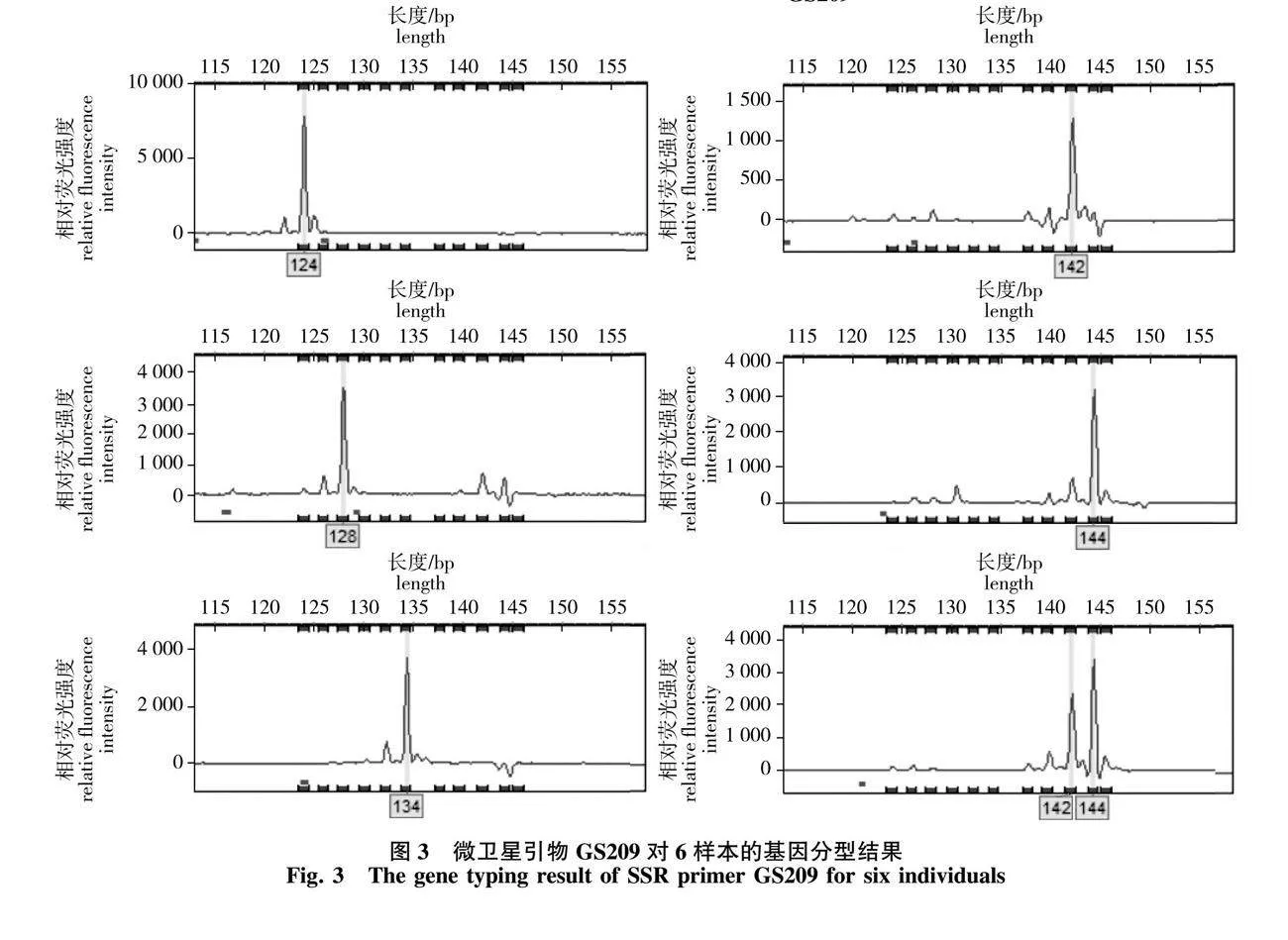
根据荧光信号,采用Genemarker V2.2.0软件对分型结果进行读取,以GS209引物为例,结果见图3。
对SSR分型结果进行读取和校对后,统计4个采集地各 12 个样本的扩增成功率。总体上,所设计的28对引物在48个样本中均具有很高的扩增率(图4)。其中,引物GS185、GS186和GS209的扩增率在所有群体中都达到了100%, GS2、GS3、GS15、GS19、GS28、GS33、GS35、GS88、GS150、GS169和GS216共11对引物的扩增率在3个群体中达到100%。从群体方面来看,云南地区的成功率均值为 83.64%,日本地区的为 91.96%,广西地区为 93.75%,江西地区为93.46%,平均扩增率为 90.7%(附表1,nldxb.njfu.edu.cn)。由此表明,这28对通用引物可以用于青冈和滇青冈跨物种的扩增。
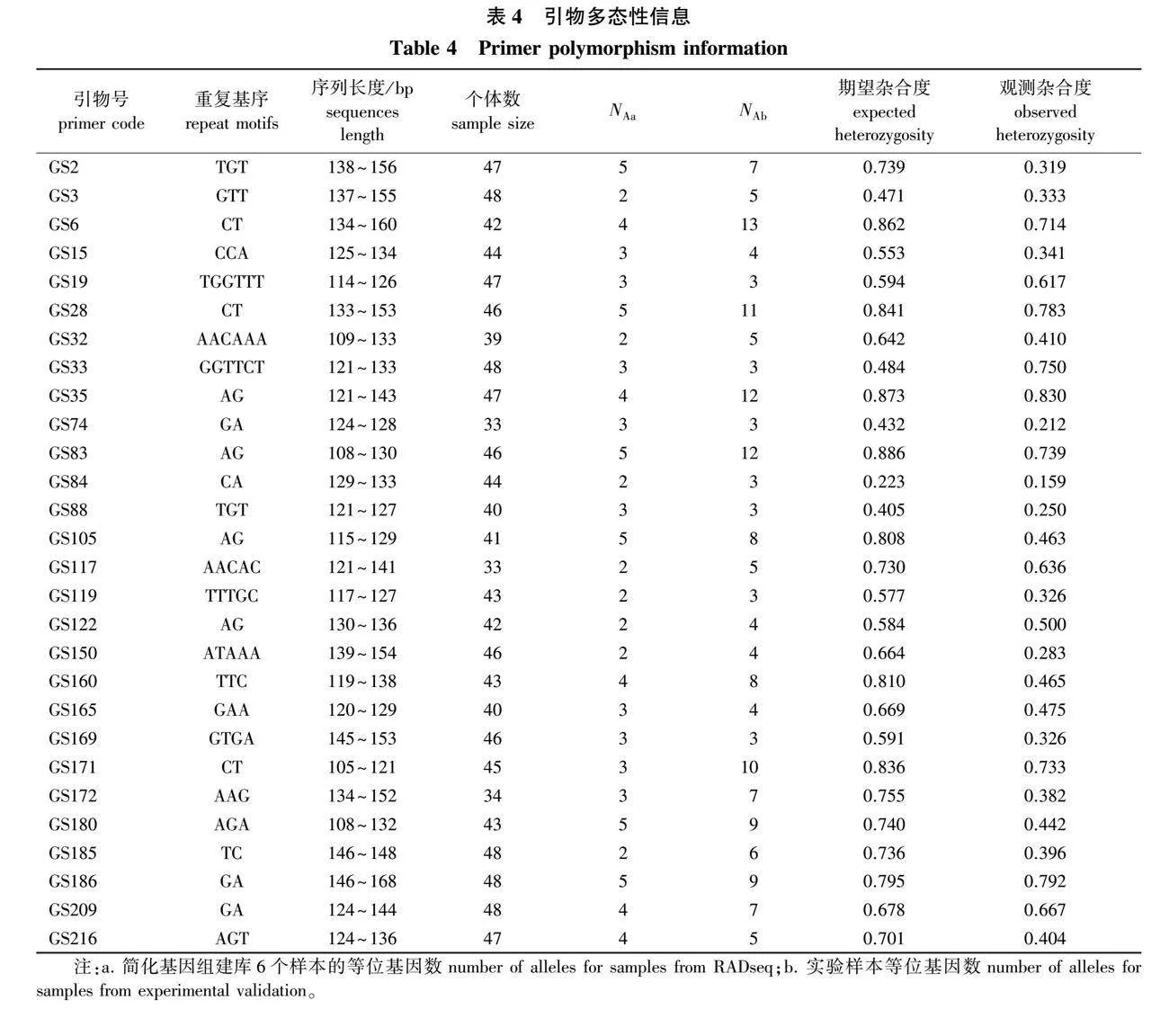
青冈和滇青冈的SSR引物多态性结果显示,基序的碱基数量在2~6均有分布,其中2碱基重复的基序最多,有12个;其次是3碱基重复的基序有9个;4碱基重复的基序最少,仅有1个。重复次数总体上随基序的碱基数的增多而减少。在所开发的28对引物中,共有176个等位基因,每个引物的等位基因数为3~13个,平均等位基因数为6.29个;期望杂合度范围为0.223~0.886,观察杂合度范围在0.159~0.830,具体结果见表4。
等位基因数是反映群体遗传变异大小的指标之一。简化基因组建库样本的等位基因数共有93个,每个引物的等位基因数为2~5个,平均为3.32个。
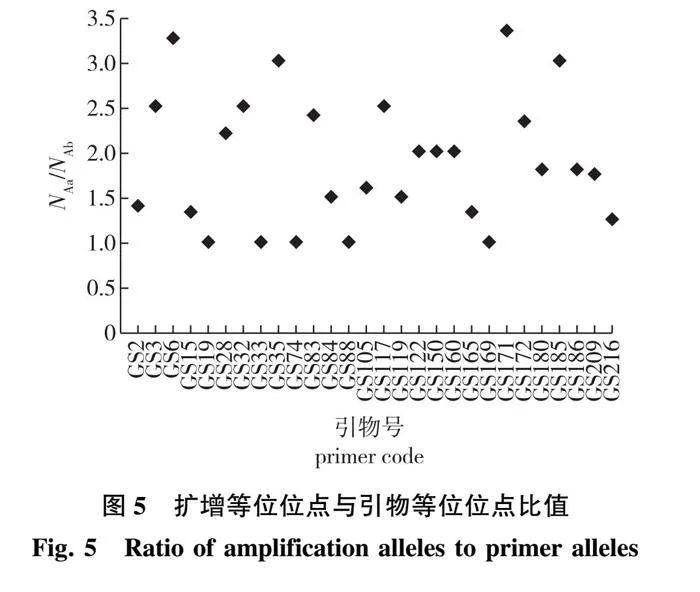
与实验样本数据相比,引物GS19、GS33、GS74、GS88和GS169所开发出的5个位点的简化基因组建库6个样本的等位基因数(NAa)与实验样本等位基因数(NAb)一致, GS2、GS3、GS15等引物所开发出的共计13个位点的NAa与 NAb基本一致(相差3个等位基因及以内)(图5)。说明这些位点的简化基因组数据不仅可用于SSR引物开发,还可用于引物的等位点数大致评估,并避免了实验的繁琐。同时,GS6、GS28、GS35、GS83和GS171这5个位点的NAa与 NAb差别很大(≥ 6),说明该位点的简化基因组数据较少,未能检测出大部分变异,故在引物筛选的时候需选择更大的样本。
3 讨 论
设计出足够数量的SSR标记是物种亲缘关系鉴定、群体遗传结构分析及保护生物学研究等方面的基础。近年来,简化基因组测序已被较多地应用于SSR引物的开发。本研究开发的青冈和滇青冈28对通用SSR引物验证结果显示,引物都能成功扩增并有多态性,引物平均等位位点为6.29。与先前基于简化基因组测序数据在其他植物SSR引物开发相比,本研究结果具有较高的成功率。如,兰进茂等[24]基于简化基因组测序开发了华蟹甲(Sinacalia tangutica)的SSR引物,在设计的200对引物中,只筛选出了20对(10%)的多态性引物;李慧等[13]开发了金银花的SSR引物,288对引物在12个金银花样本中验证显示72对(25%)引物具有多态性;Gao等[14]开发了疣柄魔芋的SSR引物,90对引物在31个个体中验证显示23对(25%)引物有多态性;宁馨等[12]开发了岭南青冈的SSR引物,25对引物在36个样本中验证显示17对(68%)引物具有多态性,引物平均等位位点为6.2。本研究能更高效地开发SSR引物的主要原因为引物开发时使用了个体间比对。通过3个青冈和2个滇青冈样本间的比对,能直观地筛选出存在变异的SSR位点及侧翼序列保守的位点,这极大地减少了实验室筛选变异SSR位点的工作量。华蟹甲、金银花和疣柄魔芋的SSR引物开发都未进行个体间的比对,显示出较低的筛选效率(lt;30%),而岭南青冈和本研究的青冈和滇青冈SSR引物开发则进行了个体间的比对,显著提高了筛选效率(gt;65%)。
总的来说,基于简化基因组测序开发SSR引物与其他方法相比有一定优势。首先,与传统的磁珠吸附方法相比,基于简化基因组开发SSR引物的方法能获得更多类型的SSR,且筛选变异SSR位点的工作量也更少。其次,近缘种筛选法和数据库搜索法虽然省略了SSR文库构建、筛选以及测序等步骤,但这两种方法的不足是已知物种SSR引物比较有限,同时由于SSR位点的侧翼序列也存在一定变异,SSR引物的种间通用性较差。第三,利用转录组序列开发SSR引物的方法最大的不足是数据量与实验量大,并且变异信息相对较少。由于转录组序列位于基因的编码区,通常具有很高的保守性,为了获得足够的遗传变异信息需要进行大批量的实验筛选,而简化基因组序列是基于全基因组DNA序列,覆盖范围更广,并且具有更高的遗传变异。同时,本研究使用了2个物种各3个样本进行样本间的聚类,能有效提高筛选出具有多态性的SSR引物的概率,减少了SSR引物后期筛选实验的工作量。此外,转录组序列开发引物对实验材料的保存要求较高,而简化基因组数据获取的材料仅用硅胶干燥保存即可,这为珍贵物种的引物开发提供了便利。最后,传统的单酶切简化基因组测序数据存在着数据利用率低、可用于分析的序列数量少、基因座精度低的缺点,本研究采用了改进的双酶切简化基因组测序技术,不但可提升测序效率,还可降低实验所需花费。
但同时,利用简化基因组开发引物也有一些不足。首先,虽然简化基因组测序一次可以获得数以万计带有SSR位点的片段,但是经过过滤、筛选后,一致性位点在本研究中仅有1 808个,最终能用于设计引物的片段更是仅有217个。其原因可能是受到了限制性内切酶的选择、测序片段长度、引物设计的要求所限制。其次,对于超大的植物基因组(基因组大小超过10G),如牡丹(Paeonia suffruticosa)[25]等,为了获取足够的一致性位点,需要测大量的原始数据,这对计算机性能等方面要求较高。最后,随着测序的迅速发展,大量全基因组的公布为开发引物提供便利,可以直接搜索并下载序列用于引物开发,省去了建库、测序的麻烦,而且还能获得更全的SSR位点信息。
基于以上存在的问题,为了更高效地开发引物,在简化基因组测序过程仍需要注意以下2点:①根据基因组大小选择合适的限制性核酸内切酶。例如,针对小基因组,需要更多的含有SSR片段的序列设计引物,因此选择酶切位点识别序列更短的酶进行酶切,如双碱基、三碱基酶切位点,具有广谱性,在基因组上的分布范围更广,酶切后能获得更多的原始序列;与之相反,若需开发超大基因组的SSR引物,则选择如六碱基这样的酶切位点识别序列更长的酶进行酶切,以减少原始序列,节省计算资源。②选择更长的测序模式。Illumina MiSeq小型测序仪经过不断优化与更新,可以一次进行250 bp(PE250)甚至300 bp(PE300)的读取。采用该方法可以获得更长的读取,从而获得更多的可用于设计引物的序列。
综上所述,简化基因组测序虽然在一些特殊情况下存在不足,但该技术有不需要物种的基因组信息、测序材料易保存、能以极低的价格获得大量均匀分布于基因组上的变异位点等优势,仍然可以进行一些近缘物种的SSR引物开发,能为杂交渐渗类群(例如栎属植物)的种群演化历史等研究打下基础,为开展群体遗传学分析提供参考。
青冈和滇青冈作为东亚亚热带常绿阔叶林重要建群种以及典型地理替代种具有很高的研究价值。本研究分别基于3个青冈和3个滇青冈植株的简化基因组数据,首先设计出28对SSR引物,然后分别在2个青冈群体和2个滇青冈群体(共48个个体)当中进行PCR扩增验证,扩增率达到了90.7%,说明该28对引物均能作为2种青冈的通用引物。从遗传多样性方面来看,在本研究所开发出的28对SSR引物中,共有176个等位基因;引物的等位基因数为3~13,平均为6.29个;引物的期望杂合度为0.223~0.886,观察杂合度为 0.159~0.830。实验结果证明,利用简化基因组测序数据,不仅可以高效、省时省力地开发引物,还可以开发近缘物种的通用引物,能为后续青冈和滇青冈的种群研究打下基础,同时也可为跨物种间的标记开发提供借鉴。
参考文献(reference):
[1]姜小龙.福建青冈和岭南青冈系统发育关系及居群遗传结构[D].长沙:中南林业科技大学,2020.JIANG X L.Phylogenetic relationship and population genetic structure of Quercus chungii and Q.championii[D].Changsha:Central South University of Forestry amp; Technology,2020.DOI:10.27662/d.cnki.gznlc.2020.000507.
[2]DENK T,GRIMM G W,MANOS P S,et al.An updated infrageneric classification of the oaks:review of previous taxonomic schemes and synthesis of evolutionary patterns in Oaks Physiological Ecology.Exploring the Functional Diversity of Genus Quercus L.[M].Cham:Springer,2017,13-38. DOI: 10.1007/978-3-319-69099-5.
[3]MANOS P S,DOYLE J J,NIXON K C.Phylogeny,biogeography,and processes of molecular differentiation in Quercus Subgenus Quercus (Fagaceae)[J].Mol Phylogenet Evol,1999,12(3):333-349.DOI: 10.1006/mpev.1999.0614.
[4]罗艳,周浙昆.青冈亚属植物的地理分布[J].云南植物研究,2001,23(1):1-16,28.LUO Y,ZHOU Z K.Phytogeography of Quercus subg. Cyclobalanopsis[J].Acta Bot Yunnanica,2001,23(1):1-16,28.
[5]中国科学院中国植物志编辑委员会.中国植物志-第四十二卷,第一分册[M].北京:科学出版社,1993.
[6]郭双兴.云南临沧晚中新世邦卖组植物群[J].古生物学报,2011,50(3):353-408.GUO S X.The late Miocene Bangmai flora from Lincang County of Yunnan,southwestern China[J].Acta Palaeontol Sin,2011,50(3):353-408.DOI: 10.19800/j.cnki.aps.2011.03.008.
[7]郭双兴.四川西部高原上新世植物群[J].古生物学报,1978,17(3):343-350,373.GUO S X.Pliocene floras of western Sichuan[J].Acta Palaeontol Sin,1978,17(3):343-350,373.DOI: 10.19800/j.cnki.aps.1978.03.007.
[8]罗冉,吴委林,张旸,等.SSR分子标记在作物遗传育种中的应用[J].基因组学与应用生物学,2010,29(1):137-143.LUO R,WU W L,ZHANG Y,et al.SSR marker and its application to crop genetics and breeding[J].Genom Appl Biol,2010,29(1):137-143.DOI: 10.3969/gab.029.000137.
[9]SELKOE K A,TOONEN R J.Microsatellites for ecologists:a practical guide to using and evaluating microsatellite markers[J].Ecol Lett,2006,9(5):615-629.DOI: 10.1111/j.1461-0248.2006.00889.x.
[10]DAVEY J W,HOHENLOHE P A,ETTER P D,et al.Genome-wide genetic marker discovery and genotyping using next-generation sequencing[J].Nat Rev Genet,2011,12(7):499-510.DOI: 10.1038/nrg3012.
[11]胡亚亚,刘兰服,冀红柳,等.简化基因组测序技术研究进展[J].江苏师范大学学报(自然科学版),2018,36(4):63-68.HU Y Y,LIU L F,JI H L,et al.Research progress on the reduced-representation genome sequencing technique[J].J Jiangsu Norm Univ (Nat Sci Ed),2018,36(4):63-68.DOI: 10.3969/j.issn.2095-4298.2018.04.012.
[12]宁馨,姜小龙,邓敏,等.基于简化基因组数据开发岭南青冈微卫星引物[J].植物研究,2020,40(4):629-634.NING X,JIANG X L,DENG M,et al.Development of microsatellite primers of Quercus championii with RAD-seq data[J].Bull Bot Res,2020,40(4):629-634.DOI: 10.7525/j.issn.1673-5102.2020.04.018.
[13]李慧,刘东超,徐瑞瑞,等.基于RAD-seq技术的金银花SSR标记开发及鉴定[J].北京林业大学学报,2021,43(6):108-117.LI H,LIU D C,XU R R,et al.Development and identification of SSR markers based on RAD-seq of Lonicera japonica[J].J Beijing For Univ,2021,43(6):108-117.DOI: 10.12171/j.1000-1522.20200337.
[14]GAO Y, YIN S, LIU C, et al. A rapid approach for SSR development in Amorphophallus paeoniifolius using RAD-seq[J]. Taiwania, 2018, (63): 281-285.
[15]王久利,陈世龙,邢睿,等.椭圆叶花锚简化基因组的SSR信息分析及SSR引物开发[J].植物研究,2018,38(2):292-297.WANG J L,CHEN S L,XING R,et al.Simplified genome SSR information and development of SSR primers of Halenia ellipitica (Gentianaceae)[J].Bull Bot Res,2018,38(2):292-297.DOI: 10.7525/j.issn.1673-5102.2018.02.018.
[16]CATCHEN J M,AMORES A,HOHENLOHE P,et al.Stacks:building and genotyping loci De novo from short-read sequences[J].G3 Genes|genomes|genetics,2011,1(3):171-182.DOI: 10.1534/g3.111.000240.
[17]KOFLER R,SCHLTTERER C,LELLEY T.SciRoKo:a new tool for whole genome microsatellite search and investigation[J].Bioinformatics,2007,23(13):1683-1685.DOI: 10.1093/bioinformatics/btm157.
[18]EATON D A R.PyRAD:Assembly of de novo RADseq loci for phylogenetic analyses[J].Bioinformatics,2014,30(13):1844-1849.DOI: 10.1093/bioinformatics/btu121.
[19]SINGH V K,MANGALAM A K,DWIVEDI S,et al.Primer premier:program for design of degenerate primers from a protein sequence[J].BioTechniques,1998,24(2):318-319.DOI: 10.2144/98242pf02.
[20]SCHUELKE M.An economic method for the fluorescent labeling of PCR fragments[J].Nat Biotechnol,2000,18(2):233-234.DOI: 10.1038/72708.
[21]HOLLAND M M,PARSON W.GeneMarker HID:a reliable software tool for the analysis of forensic STR data[J].J Forensic Sci,2011,56(1):29-35.DOI: 10.1111/j.1556-4029.2010.01565.x.
[22]PEAKALL R,SMOUSE P E.Genalex 6:genetic analysis in Excel.Population genetic software for teaching and research[J].Mol Ecol Notes,2006,6(1):288-295.DOI: 10.1111/j.1471-8286.2005.01155.x.
[23]APARICIO J M,ORTEGO J,CORDERO P J.What should we weigh to estimate heterozygosity,alleles or loci?[J].Mol Ecol,2006,15(14):4659-4665.DOI: 10.1111/j.1365-294X.2006.03111.x.
[24]兰进茂,覃瑞,夏婧.华蟹甲的简化基因组测序及SSR引物开发[J].中南民族大学学报(自然科学版),2021,40(3):258-263.LAN J M,QIN R,XIA J.Simplified genome SSR information and the development of SSR primers in Sinacalia tangutica (Asteraceae)[J].J South Central Univ Natl (Nat Sci Ed),2021,40(3):258-263.DOI: 10.12130/znmdzk.20210307.
[25]LV S Z,CHENG S,WANG Z Y,et al.Draft genome of the famous ornamental plant Paeonia suffruticosa[J].Ecol Evol,2020,10(11):4518-4530.DOI: 10.1002/ece3.5965.
(责任编辑 吴祝华)
猜你喜欢
莫愁(2023年9期)2023-03-17 09:37:12
莫愁·家教与成才(2023年3期)2023-03-15 00:55:23
特产研究(2022年6期)2023-01-17 05:05:06
四川动物(2017年6期)2017-12-12 06:14:46
四川动物(2017年4期)2017-07-31 23:54:19
魅力中国(2016年29期)2017-05-27 21:45:39
西藏科技(2016年9期)2016-09-26 12:21:38
江苏农业科学(2016年6期)2016-07-25 01:44:12
江苏农业科学(2016年6期)2016-07-25 00:16:35
中南林业科技大学学报(2015年6期)2015-12-20 05:49:35
