
拉伸速率与拉伸温度对PVDF-HFP 薄膜压电性能的影响
2024-12-09 00:00:00汪洋吴良科
重庆大学学报 2024年11期







摘要:拉伸是提高PVDF-HFP 薄膜压电性能最有效的方法之一。采用溶液浇铸法制备PVDFHFP压电薄膜,以拉伸速率和拉伸温度为变量,研究了薄膜拉伸前后形貌变化及晶体结构变化。结果表明,沿拉伸方向的应力可以迫使基体内部结构由球晶转变为纤维状晶体,从而促使非极性α 相转变成极性β 相,在拉伸伸长率为5、拉伸温度为60 ℃和拉伸速率为10 mm/min 时,薄膜的β 相相对含量超过90 %。在最大极化电场Emax=60 MV/m 作用下,其标准开环电压达到1.50 V;在此拉伸工艺下,将最大极化电场提升到100 MV/m,薄膜的标准开环电压达到2.24 V,提高最大极化电场使基体内部固有偶极矩取向更充分,压电性能更优异。
关键词:聚偏氟乙烯-六氟丙烯;拉伸;极化;晶相转变
中图分类号:TB383 文献标志码:A 文章编号:1000-582X(2024)11-094-10
PVDF-HFP 的压电性能高度依赖于极性β 相的含量与偶极矩的取向,而溶液浇铸成膜的主要晶体是热力学最稳定的非极性α 相,因此,β 相的形成以及α-β 相的转变引起了学者的广泛关注。拉伸是一种直接的诱导相变方法[1-2],然而,拉伸工艺中有较多参数,如拉伸温度、拉伸率、拉伸速率、拉伸方式(单轴或双轴)等,拉伸参数的不同往往导致薄膜性能的差异。因此,为制备高性能PVDF-HFP 压电薄膜,需要对拉伸工艺的参数进行优化。
拉伸温度是影响相变的一个极其重要的参数。Sencadas 等[3]研究了不同温度(80~140 ℃)拉伸过程中α-β 相转变,分析了在80 ℃拉伸时的应力-应变曲线,随着拉伸率增加,试样经历了屈服、颈缩和强化3 个阶段,最大应力出现在屈服硬化后和断裂前的塑性阶段,随着拉伸的进行,材料内部可以发生相变,对于较大变形,微观机制表现为从球晶结构向微纤维状晶相结构的转变,同时层状形态遭到破坏。Li 等[4]研究了不同拉伸温度、拉伸速率和拉伸伸长率下α-β 相的转变,结果表明,拉伸温度100 ℃为最佳相变温度,随着拉伸伸长率的增加,相应的结晶度也会增加,并在拉伸伸长率为3 时达到最大,而拉伸速率对相变影响较小。Debili 等[5]分析了在拉伸伸长率为4 时,不同拉伸温度对薄膜相变的影响,对比发现,80 ℃左右可诱导产生较多的β 相,聚合物在此温度下形成单β 相,当温度大于80 ℃时,诱导产生较少的β 相,并形成(α+β)双相体系。拉伸速率也是影响晶相形成的一个重要因素。Magniez 等[6]用熔融纺丝法制备了高β 相PVDF 纤维,拉伸温度保持在120 ℃,在不同拉伸速率(500、900 mm/min)和拉伸伸长率(1.25、1.50、1.75)下进行单轴拉伸。结果表明,当拉伸伸长率为1.25,拉伸速率为500 mm/min 时样品β 相含量最高,这是因为熔体应力拉伸诱导β 相形成,在低流速下产生的细丝在纺丝孔处会受到较高的熔体应力,而在高速率拉伸时,随着熔体纺丝孔聚合物流量的增加,纤维直径增加,β 相含量降低。
综上所述,拉伸工艺的参数往往能较大程度地影响薄膜β 相含量和结晶度,基于Jin 等[7]的研究,拉伸伸长率R=5.00 时,PVDF-HFP 薄膜可获得最高的β 相含量。目前对于拉伸速率和拉伸温度相关参数的研究存在较大差异,不同的工艺可能会产生不同的结果。因此,笔者采用溶液浇铸法制备初结晶薄膜后,以拉伸速率和拉伸温度为变量,探究薄膜拉伸前后形貌和晶体结构的变化,并确定拉伸工艺的最佳参数。
1 实验
1.1 材料
聚偏氟乙烯-六氟丙烯(PVDF-HFP):阿科玛公司(Arkema Inc.)产,型号为Kynar Flex 2801,熔融温度为125~164 ℃。N, N-二甲基甲酰胺(DMF):上海泰坦公司产,纯度≥99.5 % (GC)。
1.2 PVDF-HFP 压电薄膜的制备
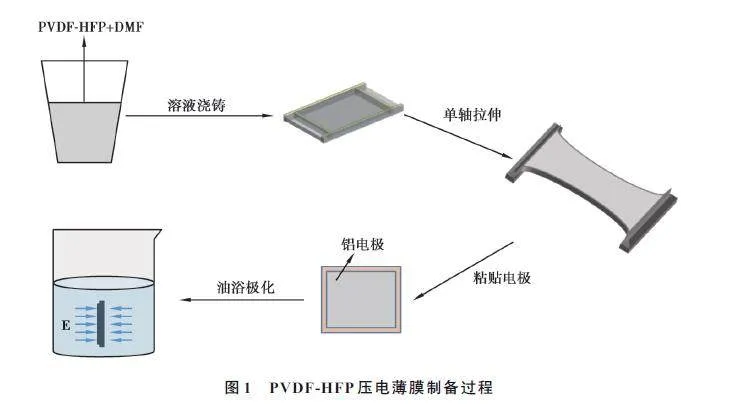
PVDF-HFP 薄膜的制备过程主要包括溶液浇铸法制膜、单轴拉伸、油浴极化3 个步骤(图1)。
1) 将8 g PVDF-HFP 粉末添加到24 g DMF 溶液中,依次对混合溶液进行行星搅拌、超声搅拌和脱泡处理,之后将混合溶液缓慢倾倒在铝板上,并在90 ℃下加热2 h,得到初结晶薄膜;
2) 为探究拉伸参数对薄膜压电性能的影响,以单轴拉伸时的拉伸温度与拉伸速率为变量,基于Jin 等[7]的研究,将拉伸率保持在5,在常温(26 ℃左右)、60、90 ℃的拉伸温度下,分别以2、5、10、15 mm/min 的拉伸速率对PVDF-HFP 薄膜进行拉伸;
3) 裁剪薄膜,尺寸为2.5 cm×3.0 cm(拉伸方向),测量薄膜厚度,用导电环氧树脂胶(ITW CW2400)将铝箔粘贴在薄膜上下表面,固化24 h,采用Step-wise 方法(分步极化法)对薄膜进行极化,最大极化电场Emax=60 MV/m。
共计12 种样品,采用统一方式对薄膜进行编号,T60 表示拉伸温度为60 ℃,下同;S2 表示拉伸速率为2 mm/min,下同;拉伸温度相对误差不超过2 ℃ ,拉伸后的压电薄膜平均厚度在50~70 μm,标准差不超过10 μm。
1.3 测试
使用聚酰亚胺(polyimide,PI)薄膜封装极化后的压电薄膜,随后,用环氧树脂胶将3 片压电薄膜粘在铝板,并连接导线,制得测试板如图2 所示(之后的表述均为测试板)。利用振动悬臂梁测试系统测试薄膜压电性能的过程如下:由正弦信号激励的电磁铁使测试板做简谐振动,使压电薄膜沿测试板长度方向产生周期性变形,振动频率为测试板的一阶固有频率(25~27 Hz),测试板末端位移为1 mm,薄膜工作模式为31 模式[8],计算各薄膜的标准开环电压Vc,以消除厚度和振幅的影响。标准开环电压可由下式计算[8]:
Vc = V(u0 t0/ut)。(1)
式中:Vc为标准开环电压,V;u 为测试板末端实际振幅,mm;t 为薄膜的平均厚度,μm;u0=1 mm 为标准振幅;t0=100 μm 为薄膜标准厚度。
1.4 表征
偏光显微镜(Carl Zeiss AG, Axio Scope A1)可以观察到薄膜的结晶形态,用以区分极性相和非极性相。
傅立叶变换红外光谱仪(Thermo Fisher Scientific, Nicolet iS50)用于测量聚合物的构象,拉伸前后薄膜的β 相相对含量F(β)按以下公式计算[8]:
式中:Aα和Aβ分别表示765 cm−1和840 cm−1的吸光度,可通过对特征峰765 cm−1和840 cm−1前后各10 cm−1的区域积分得到;Kα和Kβ分别表示α 相和β 相的吸收系数,Kα=6.1×104 cm2/mol,Kβ=7.7×104 cm2/mol。
低温差示扫描量热仪(Mettler Toledo, DSC3+)用于计算聚合物薄膜的结晶度,通过以下公式计算在不同阶段下(拉伸前、拉伸后)的压电薄膜的结晶度[8]:
Xc = (ΔHm/ΔH Φm )× 100%。(3)
式中:ΔHm 表示所测薄膜的熔融焓,J/g,可通过计算熔融峰曲线所包围的面积得出;ΔH Φm 是100 %结晶度的压电材料的理论焓值,其值为104.5 J/g。
2 结果与讨论
2.1 PVDF-HFP 薄膜的偏光显微镜(polarization microscope, POM)形貌
图3 为PVDF-HFP 薄膜拉伸前后的POM 形貌图,由图3(a)可见,未拉伸薄膜(初结晶薄膜)的基体内部以球晶层状结构为主,对应α 相。拉伸后的薄膜内部显示出不同取向程度的纤维结构,拉伸过程的微观表现是晶体结构的破坏和重组,拉伸过程中层状结构被破坏,然后在应力作用下发生结构重组,诱导纤维状β 相的形成[9]。不同拉伸温度下,纤维的取向程度有较大差异,常温下,并没有明显的纤维结构,且取向程度不规整;随着温度增加,T60 和T90 的纤维取向度显著增强,因此,在一定拉伸温度下,拉伸可诱导形成高度取向的纤维状β 相结构。
图3 中白框所圈出的黑点表示结构中空洞的形成,在其他文献中也报道了类似的情况[3,10]。当薄膜被拉伸时,层状结构被分离,形成相互连接的空洞[11],而在高速拉伸时,该情况有所改善,如图3(e)。在微观结构中,可以观察到,拉伸能有效促进纤维结构的取向,但同时也会引入缺陷,会一定程度影响薄膜的性能。
2.2 PVDF-HFP 薄膜的傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FT-IR)表征
FT-IR 光谱图在4 000 cm−1到400 cm−1的中红外区获得,图4 截取了波长范围从1 000 cm−1到600 cm−1的光谱图,分别表示在26、60、90 ℃下,以不同拉伸速率拉伸后的薄膜,图中615、765、795、975 cm−1 处的峰为α 相对应的特征峰,840 cm−1处的峰为β 相对应的特征峰[12]。
在本实验的拉伸过程中,薄膜的拉伸率统一设置为5,从图谱中可观察到,拉伸后的薄膜α 相特征峰明显减弱,β 相特征峰逐渐增强。对比各温度下不同拉伸速率下的薄膜不难发现,在常温下拉伸的薄膜,如图4(a)所示,其α 相特征峰随着拉伸速率的增加而减弱,β 相对应特征峰逐渐增强。然而,在60、90 ℃下,如图4(b)和图4(c)所示,拉伸速率对薄膜相变的影响并不显著。
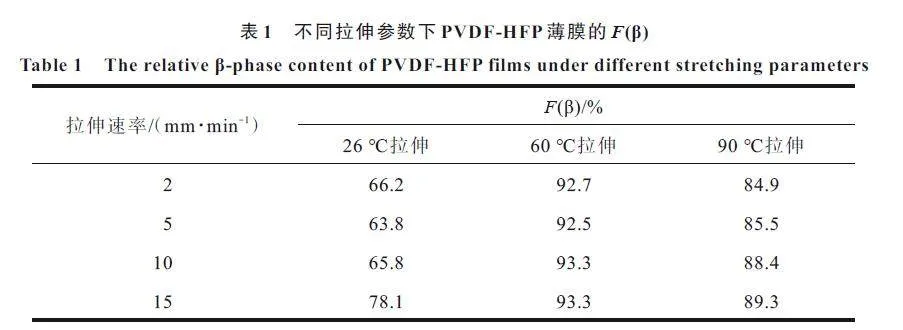
为进一步探究拉伸参数对薄膜晶相转变的影响,利用式(2)计算所有薄膜试样内部F(β),得到拉伸试样的结果如表1 所示。在室温下,未拉伸PVDF-HFP 薄膜的F(β)值为32.2%,由表1 可知,F(β)随拉伸速率的增加而增加,在15 mm/min 下β 相含量增加最显著,达到78.1%。这是因为在常温下,薄膜表现较稳定,低速率拉伸还不足以使内部球晶α 相充分转换为纤维状β 相,而当拉伸速率达到15 mm/min 时,较快的应变变化能引起晶体的充分破坏和重组,导致β 相含量升高。而在温度较高时,拉伸速率对薄膜F(β)变化影响不明显。
对比同一拉伸速率、不同拉伸温度下的薄膜发现,F(β)随温度的升高呈现先增加后减小的趋势,在60 ℃下其F(β)高达93.3%。这是因为在常温下,分子链活性较低,运动能力差,而球晶α 相转变为纤维状β 相的过程需要分子链的运动[13],因此,常温下的拉伸,晶相转变效率不高;而随着温度升高,分子链运动较活跃,且温度升高会使得薄膜延展性提高,在应力作用下,更容易促使球晶α 相转变为纤维状β 相,因此,薄膜晶相转变效率大幅提高,实现了更高的F(β);然而,当温度过高时(90 ℃),其F(β)低于60 ℃的薄膜,有报告表明,当拉伸温度低于75 ℃时,α 相转变为单β 相,而当拉伸温度超过80 ℃时,形成双晶相(α+β)体系,即拉伸温度过高,转变效率反而下降[5]。
整体而言,拉伸速率对薄膜F(β)影响较小;而拉伸温度对薄膜F(β)影响较大,拉伸温度在60 ℃时F(β)超过90%。
2.3 PVDF-HFP 薄膜的差示扫描量热仪(differential scanning calorimetry, DSC)表征
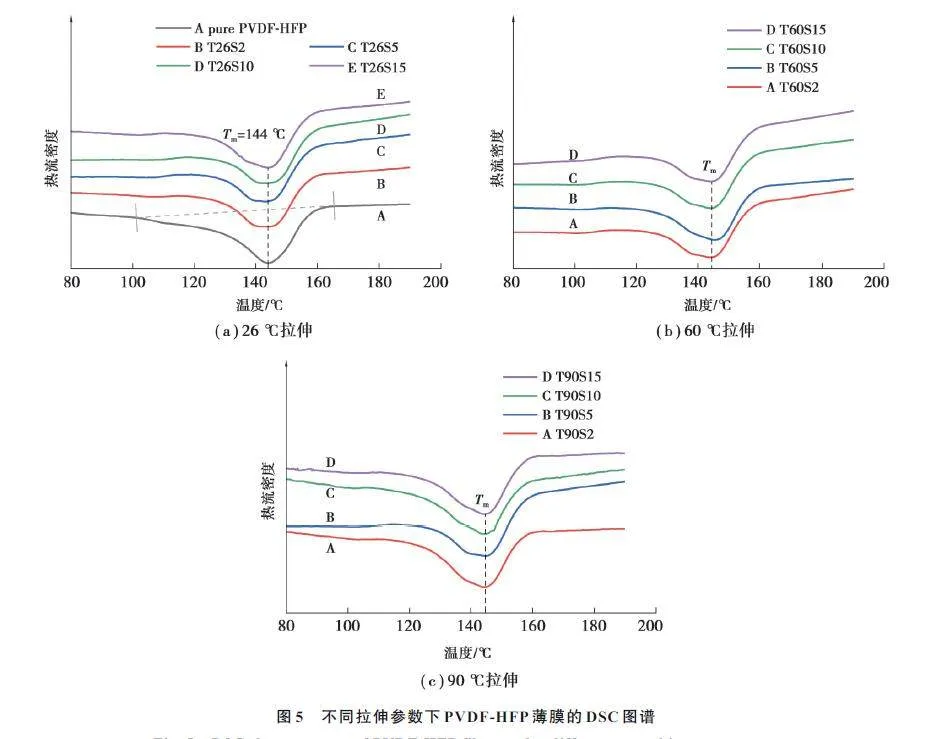
图5 显示了未拉伸薄膜以及不同拉伸工艺处理后的薄膜的DSC 曲线。温度在120 ~150 ℃ 时,PVDFHFP薄膜出现明显的熔融峰,未拉伸的PVDF-HFP 薄膜的晶体熔融峰更宽,即熔限偏大,这是因为未拉伸的薄膜内部存在更多不完善的晶体和小尺寸晶粒,导致其结晶不完善,在较低温度下开始发生熔融,造成熔限偏大[14];而拉伸后的薄膜,晶体取向程度较高,结晶更完善,因此,熔融温度Tm比较高。此外,对比未拉伸薄膜与拉伸后薄膜的曲线发现,未拉伸薄膜显示出较强的熔融峰强度,结合FT-IR 结果表明,拉伸前的薄膜主要以较稳定的α 相为主,因此,其熔融温度较为明显;而拉伸后的薄膜α+β 相共存,2 种晶相熔点不同[15],因此,熔融温度有偏移,可能造成多个熔融峰,曲线上表现为没有明显的熔融温度。
表2 列出了通过式(3)计算的不同拉伸参数下PVDF-HFP 薄膜的结晶度;未拉伸PVDF-HFP 薄膜的结晶度为38.4%;不难发现,拉伸温度对结晶度的影响较大,在常温下,薄膜的结晶度降低,其主要原因是常温下薄膜的延展性较差,分子链活性较低,此温度下拉伸会对结晶区造成破坏,导致其结晶度下降。当温度高于60 ℃时,结晶度的变化规律并不明显,结合FT-IR 结果,拉伸过程会引起聚合物基体结构的变化,沿拉伸方向取向增加,促进相变,然而在拉伸过程中,当材料达到屈服阶段后,拉伸会引入新的缺陷,从而产生内应力,导致晶片遭到破坏,内部完整性受损,因此,也可能引起结晶度的降低。在同等拉伸速率下,T60的薄膜结晶度均比T26的高,因此,60 ℃为本实验的最佳拉伸温度。
2.4 不同拉伸温度下PVDF-HFP 薄膜的应力-应变曲线分析
PVDF-HFP 作为一种热塑性高分子聚合物[16],不同温度下的拉伸对薄膜的应力-应变曲线影响较大,图6显示了不同拉伸温度下薄膜的应力-应变曲线。图6 中,R 表示拉伸伸长率,E(T26)、E(T60)、E(T90)分别表示拉伸温度为26、60、90 ℃时的杨氏模量。
由应力-应变曲线可以看出,随着应变增加,薄膜经历了弹性、屈服和强化阶段,对薄膜弹性阶段进行分析可以计算其在不同温度下的杨氏模量。图6(b)为薄膜在ε=0.00~0.03 的应力-应变曲线放大图,对该区域进行线性拟合,得到其杨氏模量,由于PVDF-HFP 是一种热塑性材料,其杨氏模量随温度变化较大,随温度的升高而降低。杨氏模量的差异说明薄膜在高温拉伸时,薄膜的延展性更好,且分子链迁移率高,因此,在不同温度下,薄膜的最大应力有较大差异。
图6(a)中标注了曲线中所对应的薄膜的拉伸伸长率,有研究表明[3,17-18],PVDF 薄膜拉伸变形初期,非晶相分子链开始伸长,晶粒开始在应力作用下取向,当拉伸伸长率R<3 时,薄膜开始由球晶结构转变为纤维状结构,并产生空洞,对应于应力-应变曲线中的弹性和屈服阶段,但在该阶段,薄膜的晶相转变并不完全;随着拉伸伸长率增加,薄膜进入强化阶段,更大的应力导致了更充分的分子链取向和伸长,诱导β 相形成,因此,该阶段对相变影响较大。对比不同拉伸温度下的曲线,常温下的曲线有较大波动,而在高温下,曲线更加光滑平整,此阶段发生塑性变形,因此,微小的波动也会导致较大的缺陷,影响相变的结果。结合晶相表征结果,当达到一定温度后(本实验为60 ℃),拉伸能更好地诱导相变。
2.5 PVDF-HFP 压电薄膜的压电性能测试
2.5.1 不同拉伸参数下PVDF-HFP 薄膜的标准开环电压测试
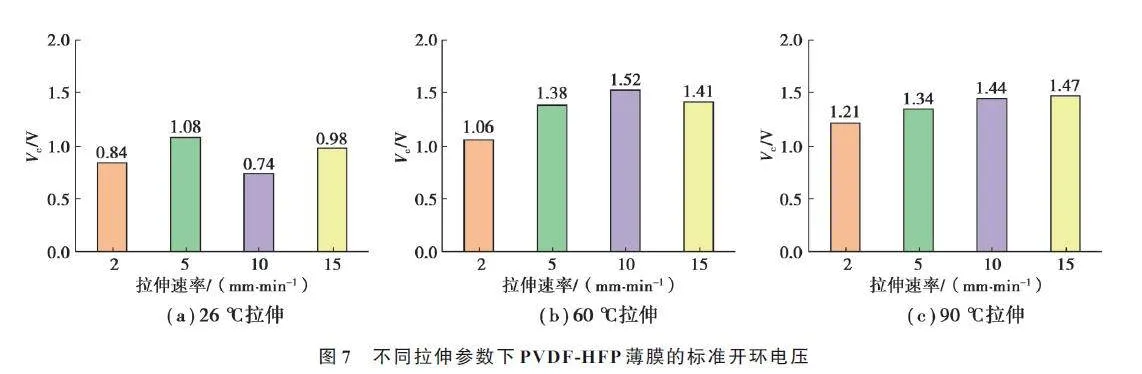
图7 中显示了不同拉伸参数下PVDF-HFP 薄膜的标准开环电压。结果表明,不同拉伸温度下,薄膜的压电性能差异较大,常温拉伸时,标准开环电压随拉伸速率变化没有明显规律,且最高标准开环电压仅为1.08 V,结合表征结果可知,在此温度下,分子链迁移率较低,且拉伸过程引入了较多的缺陷,整体结晶度降低,极性β 相含量较低,导致其标准开环电压偏低。随着拉伸温度升高,薄膜内部α-β 相转化效率大幅提高,分子链沿拉伸方向取向增加,压电性能更优异。
在60 和90 ℃拉伸时,标准开环电压随拉伸速率增加略有增加,T60S10 的开环电压达到1.52 V。拉伸速率越大,体系中的分子链排列越有序,相同极化电场强度下,可以获得更高的偶极矩取向度。拉伸速率在15 mm/min 时,标准开环电压降低可能是拉伸过程中缺陷引入导致的。整体来看,拉伸速率对压电性能的影响不大,这与晶相结构表征和结晶度分析结果一致。
2.5.2 不同极化电场下PVDF-HFP 薄膜的标准开环电压测试
通过上述实验与分析可以确定最佳拉伸参数。尽管拉伸后的薄膜具有高含量的极性β 相并且沿拉伸方向具有一定取向度,但其分子偶极矩仍是杂乱无序的,整体并不具有压电性或压电性极弱,因此,需要对薄膜进行极化以使分子链偶极矩高度取向。本节旨在进一步完善薄膜制备工艺,探究极化电场对薄膜压电性能的影响。图8 显示了不同电场强度极化下PVDF-HFP 薄膜的标准开环电压值(拉伸温度60 ℃ 、拉伸速率10 mm/min)。
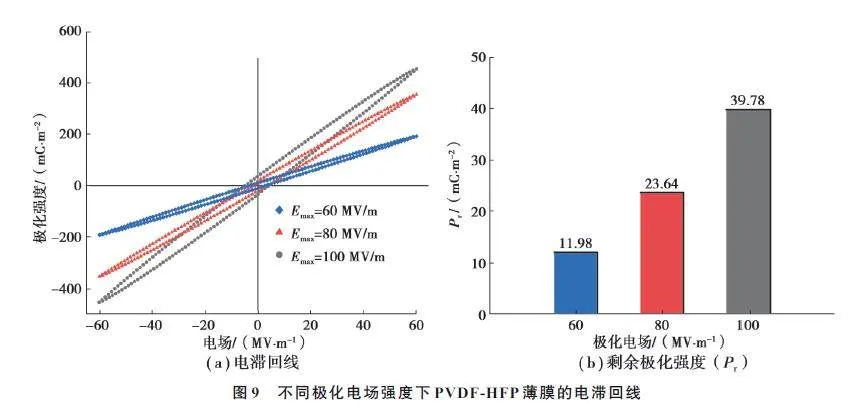
由图8 可以看出,在相同的拉伸工艺下,薄膜极化电场不同造成了薄膜压电输出的差异,极化电场越高,薄膜的标准开环电压越高,在Emax=100 MV/m 时,其标准开环电压达到2.24 V,比Emax=60 MV/m 下的薄膜电压(1.52 V)提高了47%,可见极化对薄膜的压电性能有显著影响。为进一步分析极化电场对薄膜压电性能的影响,对不同电场极化的PVDF-HFP 薄膜做了电滞回线测试,结果如图9 所示。
电滞回线是判定晶体是否为铁电体的重要依据,在交流电源下施加正弦波或三角波,每变化一个周期,便显示出电滞回线[19]。通过电滞回线可以得到剩余极化强度(Pr),即铁电体经过极化处理,撤除外电场后仍保留一定极化强度。一般情况下,剩余极化强度越大,压电性能越好。测试了不同极化电场下的薄膜在外加电场60 MV/m 下的电滞回线,随着极化电场强度的增加,Pr相应增大,从微观来讲,极化电场越高,聚合物分子偶极矩沿电场方向取向程度越高,极性越强,因此,表现出更大的剩余极化强度。其中,100 MV/m 电场强度极化下的薄膜Pr 提升至39.78 mC/m2,是60 MV/m 电场极化下的薄膜极化强度(11.98 mC/m2)的332%,其电压输出明显更高。
综上所述,理论上极化电场强度越大,薄膜剩余极化强度越高,压电输出也越高。然而,高极化电场意味着高击穿失效风险,对于纯PVDF-HFP 薄膜而言,薄膜的厚度、缺陷往往会影响极化成功率,且由于内部载流子较少,需要高电场极化才能使偶极矩充分取向。
3 结 论
研究了拉伸速率和拉伸温度对PVDF-HFP 薄膜的微观形貌、晶相结构和压电性能的影响,发现当拉伸温度高于60 ℃时,分子链取向度更加显著,但拉伸过程会使材料内部产生空洞,引起更多缺陷;拉伸温度对薄膜相变效率有较大影响,在60 ℃的拉伸温度、R=5 的拉伸伸长率下,薄膜的β 相含量超过90 %;在90 ℃时,β相含量有所下降,拉伸速率对内部相变影响不大;当拉伸温度大于60 ℃时,拉伸不会引起薄膜结晶度的下降或者影响很小,而在常温下拉伸,结晶度会下降5% 左右;标准开环电压结果表明,在60 ℃ 的拉伸温度、10 mm/min 的拉伸速率、最大极化电场为60 MV/m 时,薄膜的标准开环电压可达1.52 V;在100 MV/m 的最大极化电场下,薄膜的压电输出提升明显,从1.52 V 提升至2.24 V,说明极化对压电性能有较大的影响,其作用是使基体内杂乱的分子偶极矩沿电场方向取向,极化电场越高,取向越好,压电性能越好。
参考文献
[ 1 ] Kim G H, Hong S M, Seo Y. Piezoelectric properties of poly (vinylidene fluoride) and carbon nanotube blends: beta-phasedevelopment[J]. Physical Chemistry Chemical Physics, 2009, 11(44): 10506-10512.
[ 2 ] He F A, Lin K, Shi D L, et al. Preparation of organosilicate/PVDF composites with enhanced piezoelectricity andpyroelectricity by stretching[J]. Composites Science and Technology, 2016, 137: 138-147.
[ 3 ] Sencadas V, Gregorio R Jr, Lanceros-Méndez S. α to β phase transformation and microestructural changes of PVDF filmsinduced by uniaxial stretch[J]. Journal of Macromolecular Science, Part B, 2009, 48(3): 514-525.
[ 4 ] Li L, Zhang M Q, Rong M Z, et al. Studies on the transformation process of PVDF from α to β phase by stretching[J]. RSCAdvances, 2014, 4(8): 3938-3943.
[ 5 ] Debili S, Gasmi A, Bououdina M. Synergistic effects of stretching/polarization temperature and electric field on phasetransformation and piezoelectric properties of polyvinylidene fluoride nanofilms[J]. Applied Physics A, 2020, 126(4): 309.
[ 6 ] Magniez K, Krajewski A, Neuenhofer M, et al. Effect of drawing on the molecular orientation and polymorphism of melt-spunpolyvinylidene fluoride fibers: toward the development of piezoelectric force sensors[J]. Journal of Applied Polymer Science,2013, 129(5): 2699-2706.
[ 7 ] Jin Z N, Lei D, Wang Y, et al. Influences of poling temperature and elongation ratio on PVDF-HFP piezoelectric films[J].Nanotechnology Reviews, 2021, 10(1): 1009-1017.
[ 8 ] 吴良科. PVDF-HFP 复合材料压电性能研究[D]. 杭州: 浙江大学, 2016.
Wu L K. Research on the piezoelectricity of PVDF-HFP based composites[D]. Hangzhou: Zhejiang University, 2016. (inChinese)
[ 9 ] Guo H L, Li J Q, Meng Y F, et al. Stretch-induced stable-metastable crystal transformation of PVDF/graphene composites[J].Polymer Crystallization, 2019, 2(4): e10079.
[10] Kim T H, Jee K Y, Lee Y T. The improvement of water flux and mechanical strength of PVDF hollow fiber membranes bystretching and annealing conditions[J]. Macromolecular Research, 2015, 23(7): 592-600.
[11] Lee S. Carbon nanofiber/poly (vinylidene fluoride-hexafluoro propylene) composite films: the crystal structure and thermalproperties with various drawing temperatures[J]. Fibers and Polymers, 2013, 14(3): 441-446.
[12] Lanceros-Méndez S, Mano J F, Costa A M, et al. FTIR and DSC studies of mechanically deformed β-PVDF films[J]. Journal ofMacromolecular Science, Part B, 2001, 40(3/4): 517-527.
[13] 闫静静, 肖长发, 王纯, 等. 拉伸过程中聚偏氟乙烯纤维晶体结构变化[J]. 高分子学报, 2019, 50(7): 752-760.
Yan J J, Xiao C F, Wang C, et al. Crystalline structure changes of poly (vinylidene fluoride) fibers during stretching process[J].Acta Polymerica Sinica, 2019, 50(7): 752-760.(in Chinese)
[14] Marega C, Marigo A. Influence of annealing and chain defects on the melting behaviour of poly(vinylidene fluoride) [J].European Polymer Journal, 2003, 39(8): 1713-1720.
[15] Steinmann W, Walter S, Seide G, et al. Structure, properties, and phase transitions of melt-spun poly (vinylidene fluoride) fibers[J]. Journal of Applied Polymer Science, 2011, 120(1): 21-35.
[16] 黄楷焱. 碳纳米管/炭黑协同增强纳米复合材料的力-电效应研究[D]. 重庆: 重庆大学, 2021.
Huang K Y. Research on the force-electrical effect of synergistic enhanced MWCNT/CB composites[D]. Chongqing:Chongqing University, 2021. (in Chinese)
[17] Servet B, Broussoux D, Micheron F. Stretching induced γ to β transition in poly (vinylidene fluoride) [J]. Journal of AppliedPhysics, 1981, 52(10): 5926-5929.
[18] Sencadas V, Moreira M V, Lanceros-Méndez S, et al. α - to β transformation on PVDF films obtained by uniaxial stretch[J].Materials Science Forum, 2006, 514/515/516: 872-876.
[19] 潘柏. 铁电薄膜电滞回线的动力学标度 [D]. 南京: 南京大学, 2003.
Pan B. Dynamical scaling of ferroelectric thin film hysteresis loops [D]. Nanjing: Nanjing University, 2003. (in Chinese)
(编辑 吕建斌)
基金项目:中央高校基本科研业务费资助项目(2020CDJQY-A008)。
