
一种检测牛奶中喹诺酮类药物的高效液相色谱-串联质谱方法
2024-10-31 00:00:00韩银涛庞李艳廖柳媚陈粉娜孙洁唐涛张秋韵罗国强次仁玉珍吴凯
国外畜牧学·猪与禽 2024年5期





摘 要:本文基于高效液相色谱-串联质谱建立了一种高精密度检测牛奶中喹诺酮类药物的方法,并对所建立的方法进行了方法学的考察与验证。本研究参考已有的标准,对标准的前处理步骤进行优化,最后采用高效液相色谱-串联质谱进行分析。20种喹诺酮类药物的标准曲线相关系数为0.995 59~0.999 91,方法检出限为0.051 4~0.928 μg/kg,回收率为73.6%~110.0%,相对标准偏差为1.33%~7.69%。该方法前处理简便、高效,所得结果检出限低,回收率较好,精准度高,重复性好,可以为检验牛奶中喹诺酮类药物提供有力的方法支撑。
关键词:喹诺酮类药物;高效液相色谱-串联质谱;高精密度;高回收率
中图分类号:TS252.7 文献标志码:A 文章编号:1001-0769(2024)05-0081-08
喹诺酮类药物是一种具有抗菌谱广、抗菌力强、价格低廉等特点的人工合成抗菌药,其作用机制是抑制DNA回旋酶发挥作用,导致mRNA与蛋白质合成失控从而达到杀死细菌的目的[1]。常见的喹诺酮类药物有诺氟沙星、氧氟沙星、环丙沙星等,常用于预防和治疗畜禽细菌性疾病[2]。但这也不可避免地导致药物在动物体内残留,继而经口摄入进入人体,对人类健康产生不良影响[3]。喹诺酮类药物可对人体造成多系统的不良反应,包括胃肠道不适、中枢神经系统不良反应、肌腱炎及反应性关节炎、影响软骨发育以及肝脏损伤等[1,4]。
牛奶是日常生活中十分常见的饮品,含有丰富的蛋白质、脂肪等,具有很高的营养价值。牛奶在生产、运输、包装到销售的过程中均存在被污染的可能,从而对人体健康产生不良影响。其中抗菌药滥用导致的药物残留是不容忽视的问题,而牛奶中喹诺酮类药物的残留现象较为普遍,已成为一个严重的食品安全问题[5]。因此,建立准确测定牛奶中喹诺酮类药物残留的方法至关重要,对保障牛奶等乳产品的食品质量安全和降低健康风险具有重要意义。目前,牛奶中喹诺酮类药物的检测方法主要有免疫学方法、高效液相色谱法、超高效液相色谱法、高效液相色谱-串联质谱法(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)等[6-8]。其中高效液相色谱-串联质谱法对复杂样本的分离能力和定性效果好,可同时检测多种药物残留,具有选择性和灵敏度高的特点,应用广泛[9]。
现存大多数研究检测的喹诺酮类药物种类较少,不够全面;现行国家标准GB 29692—2013《牛奶中喹诺酮类药物残留量的测定 高效液相色谱法》也只规定了11种喹诺酮类药物的测定方法[10]。因此,有必要开发一个包括更多种类药物的测定方法。本研究以牛奶为样本,在国家标准的基础上进行优化,建立简便、快速、可重复性高的前处理方法,采用高效液相色谱-串联质谱法对样本中的依诺沙星、环丙沙星、达氟沙星、麻保沙星、恩诺沙星、沙拉沙星、二氟沙星、西诺沙星、奥比沙星、萘啶酸、噁喹酸、氟甲喹、吡哌酸、氟罗沙星、加替沙星、司帕沙星、诺氟沙星、洛美沙星、培氟沙星以及氧氟沙星这20种喹诺酮类药物进行准确的定性定量检测,以期为喹诺酮类药物的检测提供有力的方法及技术参考。
1 材料与方法
1.1 仪器
Aglient 1290超高效液相色谱仪,美国AB Sciex Triple Quad™ 5500质谱仪,OAN-EVAP 24型全自动氮吹仪,日本TAITEC Mix-VR多管漩涡混合仪,梅特勒AG285型天平,IKAKS501型旋涡混合器,德国Sigma 3K15冷冻离心机,Millipor超纯水仪,新芝SB-1200DT型超声波清洗机。
1.2 材料与试剂
乙腈(色谱纯);甲醇(色谱纯);甲酸(色谱纯);0.2%甲酸甲醇溶液(V甲醇∶V甲酸=100∶0.2);甲醇水溶液(V甲醇∶V水=1∶1)。
QuEChERS缓冲盐试剂袋;Waters PRIME HLB(6cc 200 mg)固相萃取柱;快速定量滤纸。
1.3 样本制备
量取适量牛奶,装入洁净容器作为试样,密封并标记,放置于-18 ℃冰箱中备用。
1.4 标准溶液的配制
准确称取20种喹诺酮类药物(依诺沙星、环丙沙星、达氟沙星、麻保沙星、恩诺沙星、沙拉沙星、二氟沙星、西诺沙星、奥比沙星、萘啶酸、噁喹酸、氟甲喹、吡哌酸、氟罗沙星、加替沙星、司帕沙星、诺氟沙星、洛美沙星、培氟沙星、氧氟沙星)标准品,用甲醇分别配成1 000 μg/mL的标准储备液,-20 ℃冰箱中保存,有效期 12个月;分别移取适量1 000 μg/mL的标准储备液于同一容量瓶中,用甲醇稀释成100 μg/mL混合标准溶液,-20 ℃冰箱中保存,有效期6个月;再移取100 μg/mL混合标准溶液稀释为10 μg/mL的混合标准工作液。
1.5 样本前处理
准确称取试样5.00 g于50 mL离心管中,加入0.2%甲酸甲醇溶液10 mL,涡旋混匀后振荡5 min,超声提取5 min,5 000 r/min 离心5 min,收集上清液;向上清液中加入QuEChERS缓冲盐试剂袋,充分涡旋混合1 min后,5 000 r/min离心10 min,取上清液过快速定量滤纸,所得滤液收集备用。
移取上述滤液4.0 mL,过PRIME HLB固相萃取柱洗脱,准确移取2.5 mL洗脱液至15 mL离心管内,在40 ℃水浴下氮气吹干,再用甲醇水溶液(体积比1∶1)定容至1.00 mL,涡旋后过0.22 μm滤膜,待HPLC-MS/MS分析。
1.6 HPLC-MS/MS条件
1.6.1 高效液相色谱条件
色谱柱:ACQUITYUPLC BEH C18(1.7 μm,2.1 mm×100 mm);流动相A相为0.1%甲酸水溶液;流动相B相为甲醇;柱温为40 ℃;进样量5.0 μL。梯度洗脱程序见表1。
1.6.2 质谱条件
离子源:电喷雾离子源;扫描方式:负离子扫描;检测方式:多反应监测;电喷雾电压为5 500 V;离子源温度为600 ℃;气帘气压力为28 psi;碰撞气压力为10 psi;雾化气压力为55 psi;辅助加热气压力为60 psi。
定性离子对、定量离子对、采集时间、去簇电压及碰撞能量见表2。
2 结果与讨论
2.1 标准曲线
按方法条件设置仪器参数,16种喹诺酮类药物(依诺沙星、环丙沙星、达氟沙星、麻保沙星、恩诺沙星、沙拉沙星、二氟沙星、西诺沙星、奥比沙星、萘啶酸、噁喹酸、氟甲喹、吡哌酸、氟罗沙星、加替沙星、司帕沙星)配制浓度为1.00、2.00、20.00、100.00、200.00、300.00 ng/mL的混合标准工作溶液,其余4种喹诺酮类药物(诺氟沙星、洛美沙星、培氟沙星、氧氟沙星)配制浓度为1.00、2.00、4.00、20.00、40.00、60.00 ng/mL的混合标准工作溶液,待仪器参数稳定后,对系列标准样本溶液进行测定,绘制标准曲线,相关系数r的范围为0.995 59~0.999 91。具体见表3。
2.2 方法检出限确定
以空白牛奶样本为基质,做7个平行添加试验,阳性添加20种喹诺酮类药物的浓度均为0.500 0 μg/kg,按照1.4方法进行前处理,进行上机测定,测得方法检出限范围为0.051 4 ~0.928 0 μg/kg,具体见表3。计算公式如下:
检出限(μg/kg)=K·Sb·C/X
式中:K取3;Sb为平行测试样含量的标准偏差;C为加标浓度,μg/kg;X为平行试样含量的平均值。
2.3 回收率及精密度确定
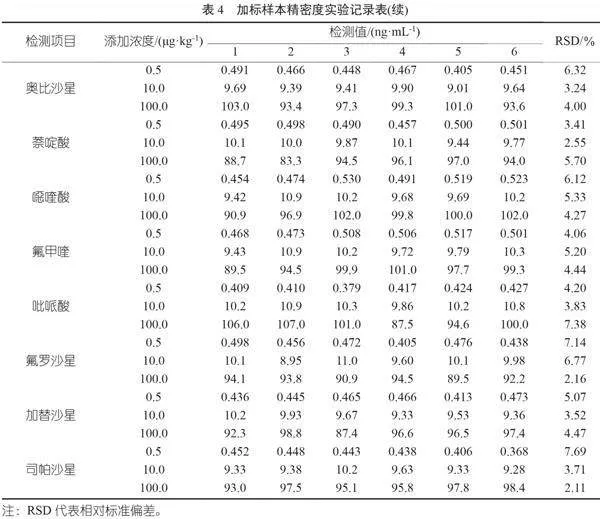
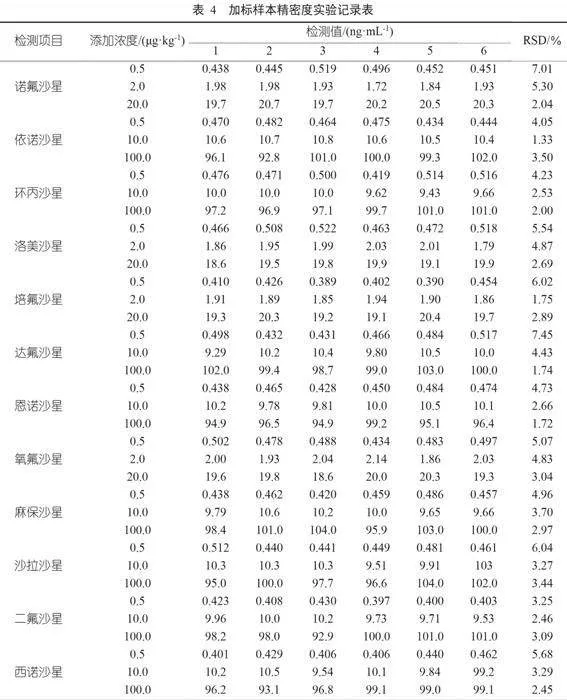
以空白牛奶样本为基质,向牛奶样本中添加16种喹诺酮类药物(依诺沙星、环丙沙星、达氟沙星、恩诺沙星、麻保沙星、沙拉沙星、二氟沙星、西诺沙星、奥比沙星、萘啶酸、噁喹酸、氟甲喹、吡哌酸、氟罗沙星、加替沙星、司帕沙星)项目混标0.5、10.0、100.0 μg/kg浓度水平,其余4种喹诺酮类药物(诺氟沙星、洛美沙星、培氟沙星、氧氟沙星)项目混标0.5、2.0、20.0 μg/kg浓度水平进行加标回收试验。按照方法进行前处理,上机测试,结果如表4及表5所示。
根据表4与表5中相对标准偏差和回收率结果可发现,该方法的相对标准偏差较低,且回收率表现较好,回收率为73.6%~110.0%,相对标准偏差为1.33%~7.69%。该方法的检出限较低,同时重复性较好,并且能保持较为优异的回收率,可用于20种喹诺酮类药物的准确定性、定量检测。
3 结论
本研究在现存标准的基础上,对牛奶中喹诺酮类药物的前处理过程进行了优化:纯化步骤采用QuEChERS缓冲盐试剂袋对样本进行初纯化,同时缓冲盐能够更好地平衡提取体系的pH和防止样本乳化;使用PRIME HLB小柱进行进一步纯化,可以进一步降低基质干扰。在高效液相色谱-串联质谱的分析中也得到了较为满意的回收率、稳定性和精密度结果。因此,本研究建立了一种基于高效液相色谱-串联质谱检测牛奶中喹诺酮类药物的简便测定方法,为相关行业检测工作提供了检测方法及技术参考。
参考文献
[1] 孙慧萍,蔡力力,阎赋琴,等.喹诺酮类药物的作用机制及不良反应[J].中华医院感染学杂志,2008,18(7):1014-1016.
[2] 李倩,王甲,张玉洁,等.动物性食品中喹诺酮类药物残留检测方法研究进展[J].食品安全质量检测学报,2021,12(8):3016-3022.
[3] 王翠月,陈大伟,马丽娜,等.动物源性食品中氟喹诺酮类药物残留现状和检测方法研究进展[J].中国家禽,2022,44(12):92-97.
[4] 李倩,张玉洁,李丹,等.兽用喹诺酮类药物的使用情况及药物残留检测进展[J].黑龙江畜牧兽医,2020(19):51-54.
[5] 肖潇,王冉冉,艾依热提·买买提,等.牛奶中6种喹诺酮类兽药残留的QuEchERS-高效液相色谱-串联质谱测定法[J].职业与健康,2023,39(8):1046-1049,1056.
[6] 张瑞婷.超高效液相串联质谱法测定牛奶中8种氟喹诺酮类兽药残留[J].食品安全导刊,2023(16):115-117.
[7] 李燕君,谭梅,岳秀英,等.牛奶中氟喹诺酮类药物残留同步快速检测的酶联免疫吸附法研究[J].中国兽药杂志,2020,54(1):46-52.
[8] 马立才,邢维维,于莹,等.牛奶中喹诺酮类药物残留的AlphaLISA检测方法的研究[J].食品工业科技,2024,45(14):264-270.
[9] 郭佩佩,刘海峰,潘明,等.牛奶中氟喹诺酮类抗生素残留检测技术现状分析[J].现代农业装备,2022,43(2):26-30.
[10] 农业部,卫生和计划生育委员会.食品安全国家标准 牛奶中喹诺酮类药物多残留的测定 高效液相色谱法:GB 29692—2013[S].北京:中国标准出版社,2014.
作者简介:韩银涛(1982- ),男,学士,高级兽医师,主要从事动物疫病预防控制相关工作;E-mail:18738294@qq.com
共同第一作者:庞李艳(1991- ),女,学士,兽医师;E-mail:348776239@qq.com
*通信作者:次仁玉珍(1994- ),女,学士,助理兽医师;E-mail: 2449552533@qq.com
吴凯(1991- ),男,学士,助理兽医师;E-mail: 465358852@qq.com
