
间伐措施对松材线虫病疫区马尾松林土壤微生物多样性的影响
2024-10-24 00:00李留彬方文马玲安一博潘超鲜李虹董智韦丹熊兴政
森林工程 2024年5期




摘 要:松材线虫病作为世界检疫性病害严重危害松属植物并威胁森林生态安全,至今没有行之有效的措施对其进行 控制和除杀。为满足人们对森林提供多种生态服务的需要,通过对马尾松林进行高强度(采伐强度60%)和低强度(采伐强 度15%)2种间伐措施采伐松材线虫病疫木后,采用高通量测序手段对松材线虫病疫区马尾松林土壤的细菌和真菌结构变化进行分析,旨在探究间伐对松材线虫病疫区马尾松林土壤微生物群落结构的变化,分析对土壤微环境的影响。采用高通量测序法对间伐抚育后的马尾松林土壤细菌和真菌群落进行研究,结果表明,3种林分土壤细菌和真菌群落表现出明显的差异,高间伐措施林分组(H-CK)的Shannon多样性指数和Chao1指数最高,H-CK组的土壤微生物群落多样性和丰富度显著高于低间伐措施林分组(L-CK)和对照组(CK)。L-CK组中标志细菌群落为Actinobacteriota(放线菌门),标志真菌群落为Leotiomycetes(锤舌菌纲);H-CK组中标志细菌群落为Chloroflexi(绿弯菌门),标志真菌群落为Dothideomycetes(座囊菌纲)。结合土壤理化性质分析表明,与对照组相比,高强度间伐H-CK组的土壤有机质含量上升13%,在增加土壤有机质积累的同时,H-CK组的全氮含量达1.12 g/kg,碱解氮的含量为64.15 mg/kg,可显著提高土壤全氮含量和有效氮含量的积累(P<0.05),从而提升林地地力。稳步提升森林质量,逐步将现有发生松材线虫病的马尾松纯林培育成复层异龄针阔混交林,使其抗病能力增加,生态功能增强。
关键词:间伐; 松材线虫病; 土壤微生物; 群落结构; 土壤化学性质
中图分类号:S763.18 文献标识码:A DOI:10.7525/j.issn.1006-8023.2024.05.009
Effects of Thinning Measures on Soil Microbial Diversity of Pinus massoniana Forest in Pine Wilt Disease Endemic Areas
Abstract: Pine wilt disease, as a world quarantine disease, seriously endangers pine plants and threatens forest ecological security. However, there are currently no effective measures to control and eliminate it. To suit people's needs for supplying diverse ecological services to the forest, high-intensity (60% logging intensity) and low-intensity (15% logging intensity) thinning measures were used in Pinus massoniana forests to harvest wood infected with pine wilt disease. The alterations in bacterial and fungal composition in the soil of P. massoniana forests impacted by the disease were analyzed using high-throughput sequencing. The objective was to investigate the alterations in the organization of the soil microbial community in P. massoniana forests impacted by thinning-induced pine wilt disease and assess the effects on the soil microenvironment. The soil bacterial and fungal populations of P. massoniana woods were investigated using the high-throughput sequencing technique. The findings demonstrated that the bacterial and fungal populations in the soil varied significantly among the three stands. The high thinning measure forest group (H-CK) had the greatest Shannon diversity index and Chao1 index, and the diversity and richness of the soil microbial community in the H-CK group were considerably higher than those in the low thinning measure forest group (L-CK) and control group (CK). Actinobacteriota was the marker bacterial community and Leotiomycetes was the marker fungus community in the L-CK group. In the H-CK group, Chloroflexi was the marker bacterial community and Dothideomycotes was the marker fungal community. The high-intensity thinning H-CK group's soil organic matter content rose by 13% as compared to the control group, according to an analysis of soil physical and chemical properties. The H-CK group's total nitrogen content increased to 1.12 g/kg and its alkaline nitrogen content was 64.15 mg/kg as soil organic matter buildup increased. The forest's soil fertility was enhanced as a result, with a significant increase in both the total and accessible nitrogen content of the soil (P<0.05). Steadily impoving forest quality, strengthening the disease resistance and boosting the ecological function of the current, pure Pinus massoniana forest that has been harmed by pine wilt disease by progressively transforming it into a multilayer, unevenly aged needle broad-leaved mixed forest.
Keywords: Thinning; pine wilt disease; soil microorganisms; community structure; soil chemical property
由松材线虫(Bursaphelenchus xylophilus)[1]引起的松材线虫病(Pine wilt disease,PWD)会对松树造成毁灭性伤害,一旦感染,松树将会迅速萎蔫死亡,同时借助媒介昆虫天牛的传播,传播扩散的速度也十分惊人[2]。1982年,在我国南京中山陵被首次发现,至今俨然成为我国松树的最大威胁,造成巨大经济和生态损失。截至2024年,该病害已扩散至我国18个省(直辖市/自治区)[3](国家林业和草原局公告[2024年第4号])。森林间伐抚育措施对于林业资源发展意义重大,影响森林生物多样性的同时,也是衡量森林质量变化的标准之一。生物多样性的多元化富集可以促进森林生态系统的优化[4],而间伐抚育则是森林经营中最主要的经营措施,间伐抚育可以改变林木生长环境,促进林木生长和林下植被发育,同时也是恢复人工林地力的重要途径[5]。间伐措施可以增加人工林物种的丰富度和地被覆盖度,研究人员发现,经过间伐抚育后的5年生和26年生的杉木林,林下空间环境因子有显著变化,而且植被生物量也得到显著增加[6]。众所周知,对于森林系统而言,生物多样性越丰富,生态系统越稳定。目前我国大部分生态公益林都处于生态敏感度低的区位,人工林组成的生态公益林又存在结构单一的问题,所以为了增加人工林的生态效益,科学抚育间伐增加生物多样性,形成更加稳定的人工林群落,才能更好地发挥出生态效益[7]。
土壤微生物作为陆地生态系统的关键组成部分[8- 9],在调节生态过程中发挥着重要作用,包括营养循环、病原体控制和植物生产力[10]。微生物群落在维持生态系统功能和服务方面,如养分循环、初级生产以及气候调节等,扮演着重要的角色[11]。土壤生物多样性的变化,主要是微生物多样性[12],可能从根本上影响生态系统功能[13]。已有研究表明,生态系统功能与物种多样性呈正相关[14],这可以归因于生态位互补性、积极的相互作用和致病物种的减少,由此说明土壤微生物在提高森林质量、增强森林抗病能力、恢复森林生态系统活力的重要性。研究人员通过对低质林进行一定程度的改造,发现20 m × 20 m的块状改造可以显著提升土壤肥力[15],同RIRhsQSR9xXfRYairA2V1yOqvD5O20pfHoYX+af1aL0=时在改造补植的过程当中,补植密度[16]和抚育间伐的强度[17]都会影响最终土壤肥力的变化。
针对目前重庆当地松材线虫病疫情发生的情况,本研究通过不同强度的间伐措施降低疫区马尾松比重,对不同间伐措施处理的松林土壤样品进行分析,探索间伐措施对林分土理化性质的影响以及土壤微生物群落结构的变化,对抚育间伐改造后的土壤微环境进行综合评价,发现高强度间伐措施(采伐强度60%)可以促进疫区马尾松林土壤微生物群落的富集,且增加林地土壤有机质含量等与提升地力相关的物质含量。可为后续重庆地区松材线虫病疫区综合改培,同时补植珍贵用材树种等相关研究提供参考和依据。
1 材料与方法
1.1 土样采集及处理
2022年9月23日,在中国重庆市梁平区星桥镇高都村(107°46′59.82″E,30°45′1.81″N)的松材线虫病防治与马尾松林综合改培研究试验区内,选取立地条件相同的林地设置试验标准样地,同时对选取样地进行间伐抚育措施。于2023年10月19日进行土样采集,采集深度为0~20 cm,分别是对照组CK(林分郁闭度0.8左右)、低强度间伐组L-CK(林分郁闭度0.6左右)和高强度间伐组H-CK(林分郁闭度0.1左右),试验组抚育强度分别为CK组(0%)、L-CK组(采伐强度15%)、H-CK组(采伐强度60%),每组设6个生物学重复。将采集好的土样分成 2份,放置在-80 ℃冰箱里一份待测土壤微生物群落变化,一份待测土壤理化性质。
1.2 DNA提取和扩增子测序
使用OMEGA公司的DNA试剂盒提取总基因组DNA样本(M5635-02)(OmegaBio-Tek,Norcross,GA,USA),提取方法根据试剂盒的说明。分别使用NanoDrop NC2000分光光度计(Thermo Fisher Scientific,USA)和琼脂糖凝胶电泳检测提取的DNA的质量浓度和质量。细菌16S rRNA的V5—V7区域的测序使用特异性引物扩增,具体引物为:799F(5′- AACMGGATTAGAGATCKG-3′)和反向引物1193R(5′-ACGTCATCCCCACCTTCC-3′);真菌ITS区域的测序使用特异性引物扩增,具体引物为:ITSF(CTTGGTCATTTAGAGGAGGAAGTAA)和ITS2(GCTGCGTTCTTCATCGATGC)。聚合酶链反应(Polymerase Chain Reaction,PCR)组分包含5 μL缓冲液(5×)、0.25 μL Fast pfu DNA聚合酶(5 U/μL)、2 μL(2.5 mM)dNTPs、1 μL(10 μM)每个正向和反向引物、1 μL DNA模板和14.75 μL ddH2O。热循环包括在98 ℃下初始变性5 min,然后是25个循环,包括在98 ℃下变性30 s,在53 ℃下退火30 s,以及在72 ℃下延伸45 s,72 ℃下最后延长5 min。PCR产物纯化后,送至上海派森诺生物科技有限公司使用Illumina NovaSeq平台和NovaSeq6000 SP试剂盒(600个循环)进行对端2×250 bp测序。
1.3 测序原始数据处理
测序得到的数据使用QIIME2 2019.4[18]进行分析,根据官方教程(https://docs.qiime2.org/2019.4/tutorials/)进行一些修改。使用解复用插件对原始序列数据进行解复用,然后使用cutadapt插件切割引物[19]。使用DADA2插件进行原始数据质量过滤、去噪、合并和去除嵌合体[20],利用mafft对齐分类单元进行分类[21]。根据SILVA版本132/UNITE版本8.0数据库,使用特征分类器插件[22]中的分类,分配给分类操作单元(Operational Taxonomic Units,OTU)或扩增子序列变异(Amplicon Sequence Variants,ASVs)。
1.4 数据分析方法及软件
序列数据分析主要使用QIIME2和R软件包(v3.2.0)进行。OTU水平的α多样性指数,如Chao1丰富度估计值[23]、Shannon多样性指数[24-25]、Simpson指数[26]和Pielou均匀度指数[27],使用QIIME2中的OTU表进行计算,并可视化为柱状图。生成OTU水平排序的丰度曲线,以比较样本间OTU的丰富度和均匀性。使用Jaccard指标[28]、Bray-Curtis指标[29]和UniFrac距离指标[30]进行β多样性分析,以调查样本间微生物群落的结构变化,并通过主坐标分析(principal co-ordinates analysis,PCoA)进行可视化。主成分分析(Principal Components Analysis,PCA)是基于属级组成的剖面计算[31]。基于样本/组中OTU的出现情况(无论其相对丰度如何),使用R包“VennDiagram”生成Venn图,以可视化样本或组之间的共享和唯一OTU。使用QIME2和默认设置,应用随机森林分析来区分不同组的样本[32- 33]。利用非加权组平均法(unweighted pair-group method with arithmetic means,UPGMA)分析微生物多样性。
1.5 土壤基本理化性质分析
土壤化学分析采用玻璃电极法测定pH、水土比5∶1v/m,元素分析仪(ELEMENTAR Vario El Cube)通过干式燃烧分析土壤有机碳(Organic Carbon,SOC)和土壤全氮Z07VWFolnBRvW1/1jiZLrZM4axJwijTbDPckW+ReUCU=(Total Nitrogen,TN),碱解扩散法测定土壤碱解氮(Alkaline Hydrolysis Nitrogen,AN)含量,火焰光度计法测定土壤NH4OAc(乙酸铵)提取液中有效钾(Available Potassium,AK)含量,钼锑抗比色法测定土壤盐酸-氟化铵浸提液中有效磷(Available Phosphorus,AP)含量。
2 结果与分析
2.1 微生物群落的丰富度及富集分析
为保证各样本在同一测序深度水平下进行进一步分析,抽平后CK组(对照)、L-CK组(低间伐)、H-CK组(高间伐)分别获得土壤细菌的有效序列为535、460、489条序列;分别获得土壤真菌的有效序列为106、69、84条序列。由图1可知,Chao1指数 可表征土壤微生物群落的丰富度,结果表明,CK组的细菌(图1(a))和真菌(图1(b))丰富度均与处理组没有显著性差异,而L-CK组与H-CK组之间的细菌群落(图1(a))和真菌群落(图1(b))丰富度有显著差异(P<0.05),同时H-CK组中细菌与真菌的Chao1指数均高于L-CK组,说明H-CK组的土壤微生物群落丰富度高于其他2组。Shannon指数和Simpson指数用于综合考虑确定组间微生物群落多样性,关于细菌多样性(图1(a)),H-CK组Shannon指数最高,并与其他2组有显著性差异(P<0.05); L-CK组Simpson指数最高,与CK组有显著差异(P<0.05)。而对于真菌多样性(图1(b)),CK组Shannon指数和Simpson指数均高于其他2组。Pielou均匀度指数用于确定均匀性,细菌群落均匀性H-CK组Pielou指数最高且与其他2组有显著差异(P<0.05);而真菌群落均匀性CK组Pielou指数最高。由此说明,经过不同程度间伐处理后,马尾松林土壤微生物的细菌多样性及丰富度均有提升,真菌多样性及丰富度呈下降趋势,间伐措施对细菌群落影响较大。
2.2 微生物组成的多样性及组成分析
基于Jaccard指标的PCoA分析结果,不同处理类型马尾松林土壤之间的群落组成差异明显,如图2(a)和图2(b)所示。根据细菌门水平分类结果,16S rDNA测定序列被归类为10个不同的门,如图2(c)所示。3种处理的马尾松林土壤细菌群落总体呈现出相似的门水平分类,但相对丰度存在差异。结合表1可知,第一,最丰富的酸杆菌门(Acidobacteria)在CK组和L-CK组样本中均占1/3以上,但在H-CK组的样品中的占比有所下降,只有24%。第二,优势门变形菌门(Proteobacteria)平均相对丰度为29%,CK组样品中的占比略高于其他2组样品,但在3组土壤中的相对占比没有显著差异。第三,优势门放线菌门(Actinobacteria)和第4优势门绿弯菌门(Chloroflexi)在H-CK组中的占比分别为18.45%和15.51%,显著(P<0.05)高于CK组和L-CK组中的占比。根据真菌门水平上前5个分类结果,对照组CK的真菌群落相对丰度与其他两者差异不大,如图2(d)所示。结合表1可知,子囊菌门(Ascomycota)和担子菌门(Basidiomycota)为3组土壤样品中的优势真菌门类,且3组真菌相对占比没有显著差异。值得注意的是,在H-CK组中毛霉门(Mucoromycota)占比达0.7%,与其他2组样品差异显著。
2.3 关键微生物群落的筛选
对CK组、L-CK组和H-CK组3组样品的差异群落进行分析,3组样品之间共同拥有301种细菌群落和69种真菌群落,如图3(a)和图3(b)所示。表明间伐处理措施影响马尾松林土壤微生物群落差异变化主要集中在细菌群落。同时,利用线性判别分析(Linear discriminant analysis Effect Size,LEfSe)对3组样品种的差异群落分析后发现,CK组中造成组间差异的标志细菌群落为拟杆菌门(Bacteroidota),真菌群落为担子菌门(Basidiomycota);L-CK组中标志细菌群落为放线菌门(Actinobacteriota),标志真菌群落为锤舌菌纲(Leotiomycetes);H-CK组中标志细菌群落为绿弯菌门(Chloroflexi),标志真菌群落为座囊菌纲(Dothideomycetes),如图3(c)和图3(d)所示。
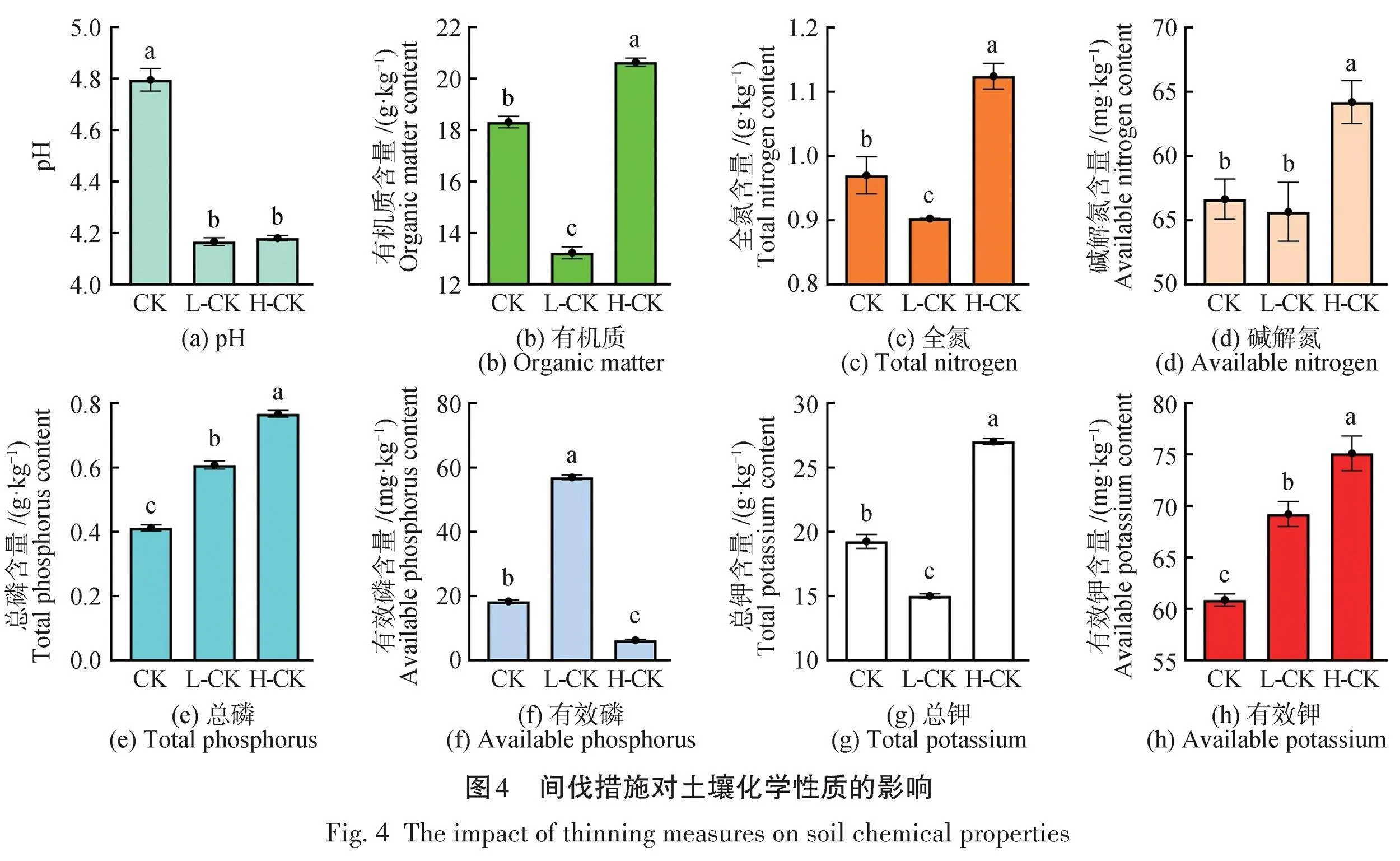
2.4 理化性质
间伐处理显著降低了土壤pH,与对照处理CK(pH=4.97)相比,低强度间伐处理L-CK(pH=4.16)和高强度间伐处理H-CK(pH=4.18)土壤pH显著 (P<0.05)下降,如图4(a)所示;CK、L-CK、H-CK土壤有机质含量分别为18.31、13.22、20.64 g/kg,如图4(b)所示;土壤的全氮含量分别为0.97、0.90、1.12 g/kg,如图4(c)所示;总钾含量分别为19.24、14.99、27.01 g/kg,如图4(g)所示。土壤有机质、全氮和总钾含量在3个处理中均存在显著差异,低强度间伐组含量最低,而在高强度间伐组含量最高,与对照组相比,低强度间伐土壤有机质、全氮和总钾含量分别显著降低了28%、7%和22%,高强度间伐组分别显著增加了13%、15%和40%。说明土壤有机质、全氮和总钾含量对间伐强度的响应机制存在差异。CK、L-CK、H-CK土壤总磷含量分别为0.41、0.61、0.76 g/kg,如图4(e)所示;有效钾含量分别为60.84、69.21、75.11 mg/kg,如图4(h)所示;两者在土壤中的含量均随着间伐强度的上升而显著增加,与对照组相比,土壤总磷含量在低强度间伐和高强度间伐处理中分别增加了48.78%和85.37%;土壤有效钾含量分别增加了14%和22%。对0~20 cm土层的土壤来讲,低强度间伐会降低土壤有机质,而高强度间伐会增加土壤有机质的积累或储存;解氮的含量分别为56.62、55.65、64.15 mg/kg,如图4(d)所示。值得注意的是,H-CK组中土壤中的全氮含量与碱解氮(水解性氮)都显著高于其他 2组(P<0.05)。与CK组对比,全氮含量提升了15%;碱解氮含量提升了13%。说明高强度间伐措施可提高土壤全氮含量和有效氮含量的积累。
由图4(f)可知,CK、L-CK和H-CK的有效磷的含量分别为18.43、57.04、6.36 mg/kg。与CK组对比,L-CK组全磷含量提升了49%、H-CK组提升了85%,达到显著差异(P<0.05);L-CK组的有效磷含量提升了209%,H-CK组降低了68%,达到显著差异(P<0.05)。说明低强度间伐措施对土壤有效磷的含量影响巨大;由图4(g)和图4(h)可知,CK、L-CK、H-CK的总钾含量分别为19.24、14.99、27.01 g/kg;有效钾的含量分别为60.84、69.21、75.11 mg/kg。与CK组对比,L-CK组总钾含量降低了22%、H-CK组提升了40%,达到显著差异(P<0.05);L-CK组有效钾含量提升了14%,H-CK组提升了23%,达到显著差异(P<0.05)。说明高强度间伐措施可提高土壤总钾含量和有效钾含量的积累。
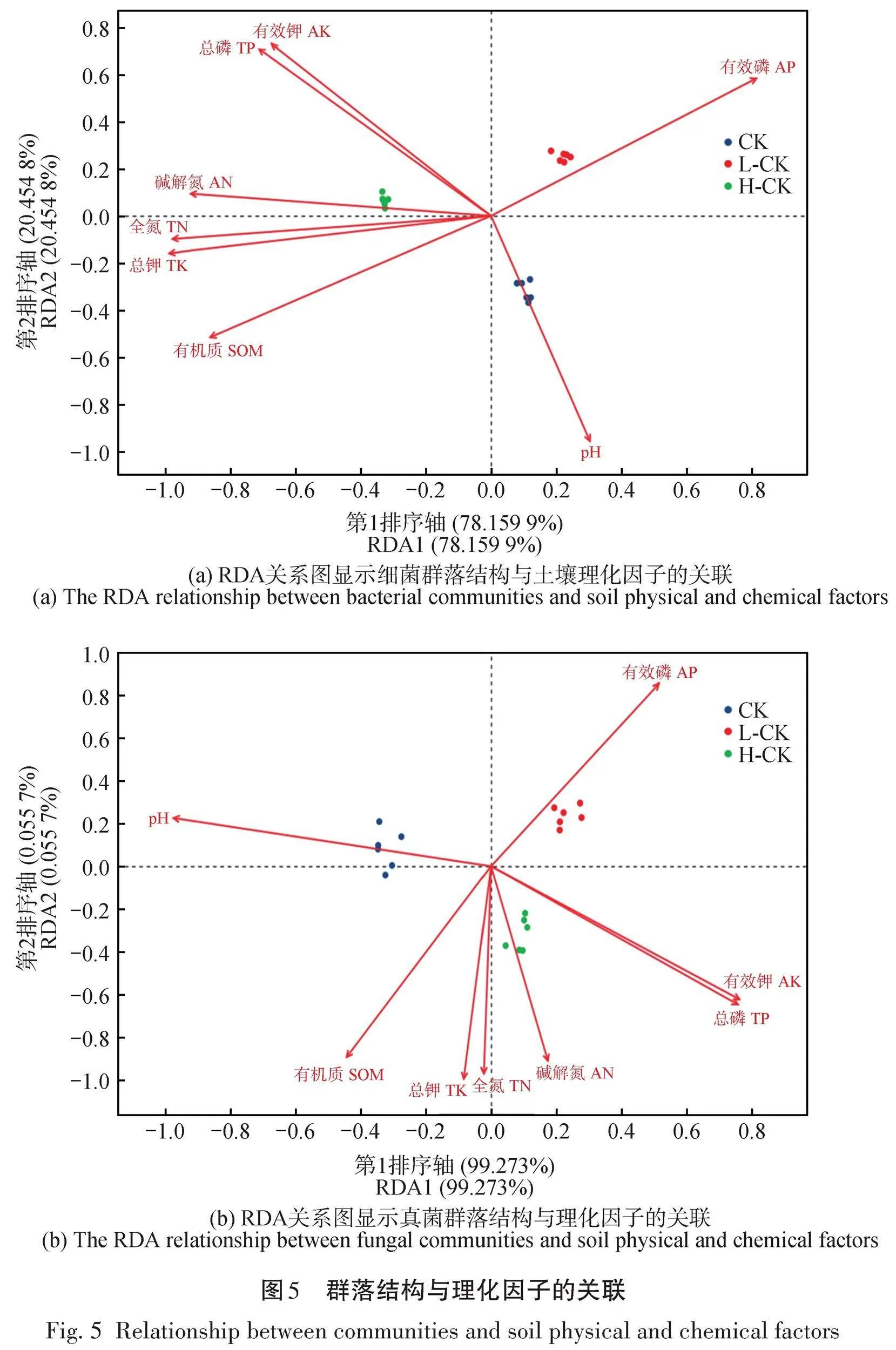
2.5 微生物群落与环境因子相关性
为研究经过不同强度间伐措施后土壤环境对细菌群落结构的影响,将土壤化学性质(pH、有机质、全氮、全磷、全钾、碱解氮、有效磷、有效钾)以及微生物群落组成进行冗余分析(Redundancy analysis,RDA),这样可以更为直观清楚地反映环境因子对土壤微生物群落结构的影响。由图5(a)可知,在各个土壤理化性质中,全钾射线最长,其次为全氮,全钾与全氮对土壤细菌群落影响程度最高,pH射线最短,其次是有效钾,pH和有效钾对土壤细菌群落影响程度最低;H-CK组分布于RDA1坐标的负方向,CK组和L-CK组主要分布于RDA1坐标的正方向,CK组和L-CK组分布于RDA2坐标的正方向,H-CK组分布于RDA2坐标的负方向。彼此之间分布都有一定的距离,说明经过间伐处理后各组土壤细菌有显著变化,且各具特点不再相似。由图5(b)可知,pH射线最长,其次为全磷,pH与全磷对土壤真菌群落影响程度最高,碱解氮射线最短,其次是全氮,碱解氮和全氮对土壤真菌群落影响程度最低;CK组分布于RDA1坐标的负方向,L-CK和H-CK分布于RDA1坐标的正方向,CK组和L-CK组分布于RDA2坐标的正方向,H-CK组分布于RDA2坐标的负方向。与细菌群落相比,影响真菌群落的环境因子有所不同,但总体分布还是有一定距离。
3 讨论与结论
“植被-土壤-微生物”作为一个整体系统,其内在的协同关系是植物群落受到干扰后研究的关键。土壤微生物是土壤有机质和养分的储备库,在森林生态系统中对土壤养分转化与循环、病菌抑制、植物生长调控以及初级生产力方面发挥着关键性的作用[34]。森林间伐是世界范围内广泛应用的林业经营实践之一,有研究表明,不同的间伐强度可引起林下植物、土壤理化性质和土壤微生物等多种多样的生态响应[35]。植物和微生物在进化中一直处于相互影响、相互制约的状态,同一植物病原体可能在不同的植物中发挥积极或消极的作用[37]。在松材线虫侵染松树的过程中,松树中微生物群落的组成将发生变化。一些特定微生物的丰度最终会影响松树的健康[38]。前人研究发现,对线虫有毒的细菌主要是芽孢杆菌属[39]和沙雷氏菌属[40]的种类,真菌主要是木霉属[41]和Esteya属[42]的种类。基于高通量分析结果表明,3种林分土壤细菌和真菌群落表现出明显的差异,L-CK和H-CK土壤微生物多样性和群落结构均与CK存在显著差异。从alpha多样性的分析结果来看,H-CK组的土壤微生物群落丰富度高于其他2组(P<0.05),且Chao1指数均为H-CK最高、L-CK组最低(P<0.05),由此表明马尾松林经高间伐措施处理后,土壤细菌群落分布均匀且丰富度提升;多样性指数结果表明,真菌多样性及丰富度呈下降趋势,高间伐措施对土壤真菌具有选择作用,间伐措施改变生境内真菌群落的栖息环境且更加适宜细菌生长,某些细菌群落成为优势种群,抑制真菌群落的繁殖生长,导致土壤真菌总体多样性下降。
间伐处理后从3种林分的土壤物种组成看,虽然土壤细菌群落的主要优势门类一致,组间的优势门的相对比例明显不同(表1)。值得注意的是,通过线性判别分析3组样品中的差异群落,如图3(c)和图3(d)所示。L-CK组中标志细菌群落为Actinobacteriota,且此类细菌种群所占比例只有0.08%,显著低于其他2组。推测间伐措施改变土壤微生物群落构成,使得某些群落成为优势种群同时发挥一定作用,造成土壤养分发生变化。Actinobacteria在自然界存在广泛[43],多数为腐生型,且其在维持生态系统物质循环[44]与加速有机物分解,促进土壤养分转化都有积极作用[45]。间伐措施改变了土壤中放线菌的比例,所以造成H-CK组的土壤有机质含量均高于其他2组,如图4(b)和图4(f)所示。放线菌在生长过程中,可代谢多种物质如茴香霉素、放线菌酮和多氧菌素等,这些物质可以抑制多种病原微生物,刺激诱导植物免疫,增强其抗病能力,提升森林生态系统稳定性[46];与此同时,H-CK组土壤绿弯菌门(Chloroflexi)相对丰度高达18%,显著高于其他2组,推测高间伐措施使得马尾松林土壤的绿弯菌门微生物富集,从而影响土壤理化性质的差异变化。经过土壤检测(图4),发现除有效磷含量与pH读数低于其他2组,其余6项土壤指标均显著高于其他2组(P<0.05)。绿弯菌的分布广泛,在深水湖泊中,绿弯菌的相对丰度高达26%[47]。对于绿弯菌门的富集,有研究人员认为绿弯菌门相对丰度占比高可能是不良群落结构的特征,如连作后的人工杨树林[48]绿弯菌门微生物的相对丰度显著升高,且连作土壤的营养代谢与绿弯菌门的富集有较强的负相关性,致使烤烟生长受阻[49],因此绿弯菌的富集可能导致土壤环境不利于马尾松林的生长;相反,绿弯菌因其独特固碳的方式“3-HP固定CO2”和丰富的代谢途径,参与C[50]、N[51-52]和硫(S)[53]等一系列重要生源元素的生物地球化学循环过程,对于森林的固碳能力和土壤养分的提升有一定的贡献。针对高强度间伐后的马尾松林,相关微生物的富集是否有利于接下来的森林生产经营,还需进一步开展营林措施后,验证其在提升森林生态效益的作用。
抚育间伐对松材线虫病疫区马尾松林的土壤理化特征有显著的影响。土壤pH是土壤各种化学性质的综合体现,研究发现,间伐处理显著降低了土壤pH,这可能是由于间伐后林内温度上升,促进了微生物对凋落物的分解,从而使凋落物中的有机酸降低了土壤pH[54]。不同的土壤pH可以影响土壤有机质的合成及分解、土壤微生物的活动与种类等,见表1和图2。土壤有效钾和总磷含量随着间伐强度的上升而显著增加,这可能是由于间伐后改善了林内光照和土壤环境,从而大幅降低了植物对资源的竞争,在林下更新先锋种未大面积建立起来前实现了土壤有效钾和总磷含量的冗余。本研究发现,与对照组相比,高强度间伐显著提高了土壤有机质、全氮和碱解氮、总磷、总钾和有效钾的含量,但对轻度间伐的正向影响却不大,这可能是由于间伐地表凋落物,凋落物中的养分没有参与到植物-土壤-微生物的养分循环中,而高强度间伐后降低了大幅林分密度,同时高强度抚育改变林内光照、湿度和郁闭度等气候条件,对有益微生物,如硝化细菌、固氮菌和氨化细菌的富集创造了条件,促进其生长及活动,这样不仅加快土壤有机质的分解效率,使得有机质含量有显著提升,如图4(b)所示。这同样与本研究土壤微生物的研究结果相符合,贫营养型细菌酸杆菌门在CK和L-CK组中占比均超过1/3,在H-CK组中下降到不到1/4[55]。高明等[56]得出19%~21%中等强度抚育间伐方式有利于土壤化学性质改善。结合本研究的结果,60%高强度抚育间伐方式对土壤化学性质的改善更加明显,同时增加有机质含量、富集有益细菌、增强土壤的固碳能力。至于19%~21%中等强度抚育间伐是否更加适合马尾松林,还需进一步试验说明,后续土壤肥力的变化和具体营林措施的影响情况也需要长期的科学监测和分析。
【参 考 文 献】
[1] NICKLE W R,GOLDEN A M,MAMIYA Y,et al.On the taxonomy and morphology of the pine wood nematode,Bursaphelenchus xylophilus (Steiner & Buhrer 1934) Nickle 1970[J].Journal of Nematology,1981,13(3):385-392.
[2] 叶建仁.松材线虫病在中国的流行现状、防治技术与对策分析[J].林业科学,2019,55(9):1-10.
EDW9D+/xZX21nsOn9hKjjQ==YE J R.Epidemic Status of pine wilt disease in China and its prevention and control techniques and counter measures[J].Scientia Silvae Sinicae,2019,55(9):1-10.
[3] 国家林业和草原局.国家林业和草原局公告(2024年第4号)(2024年松材线虫病疫区)[EB/OL].(2024-02-19)[2024-05-12].https://www.forestry.gov.cn/search/547482
National Forestry and Grassland Administration Government Network.Announcement of the National Forestry and Grassland Administration (No.4 of 2024) (2024 pine wood nematode disease epidemic area)[EB/OL].(2024-02-19)[2024-05-12].https://www.forestry.gov.cn/search/547482
[4] 陈贵兰.试析生物多样性保护——以福安市为例[J].林业建设,2020(5):54-58.
CHEN G L.Analysis on biodiversity conservation: a case study of Fu′an city[J].Forestry Construction,2020(5):54-58.
[5] 李春明,杜纪山,张会儒.抚育间伐对森林生长的影响及其模型研究[J].林业科学研究,2003(5):636-641.
LI C M,DU J S,ZHANG H R.The effects of thinning on forest growth and model study[J].Forest Research,2003(5):636-641.
[6] 熊有强,盛炜彤,曾满生.不同间伐强度杉木林下植被发育及生物量研究[J].林业科学研究,1995(4):408-413.
XIONG Y Q,SHENG W T,ZENG M S.A study on the development and biomass of undergrowth vegetation in Chinese fir plantation with different thinning intensities[J].Forest Research,1995(4):408-413.
[7] 祝顺万,刘利霞,胡雪凡,等.华北落叶松混交林林下植物群落特征对间伐的响应[J].森林工程,2024,40(3):47-55.
ZHU S W,LIU L X,HU X F,et al.The effects of different thinning intensities on the understory vegetation characteristics of mixed forests of Larix principis-rupprechtii[J].Forest Engineering,2024,40(3):47-55.
[8] WANG C,WANG X,LIU D,et al.Aridity threshold in controlling ecosystem nitrogen cycling in arid and semi-arid grasslands[J].Nature Communications,2014,5:4799.
[9] 李琳,杜倩,刘铁男,等.松嫩平原植被演替对土壤微生物的影响[J].森林工程,2022,38(4):45-52.
LI L,DU Q,LIU T N,et al.Effects of vegetation succession on soil microorganisms in Songnen Plain[J].Forest Engineering,2022,38(4):45-52.
[10] HAN S,TAN S,WANG A,et al.Bacterial rather than fungal diversity and community assembly drive soil multifunctionality in a subtropical forest ecosystem[J].Environmental Microbiology Reports,2022,14(1):85-95.
[11] DELGADO-BAQUERIZO M,MAESTRE F T,REICH P B,et al.Microbial diversity drives multifunctionality in terrestrial ecosystems[J].Nature Communications,2016,7:10541.
[12] DELGADO-BAQUERIZO M,ELDRIDGE D J,OCHOA V,et al.Soil microbial communities drive the resistance of ecosystem multifunctionality to global change in drylands across the globe[J].Ecology Letters,2017,20(10):1295-1305.
[13] YANG G,RYO M,ROY J,et al.Multiple anthropogenic pressures eliminate the effects of soil microbial diversity on ecosystem functions in experimental microcosms[J].Nature Communications,2022,13(1):4260.
[14] DELGADO-BAQUERIZO M,GUERRA C A,CANO-DÍAZ C,et al.The proportion of soil-borne pathogens increases with warming at the global scale[J].Nature Climate Change,2020,10(6):550-554.
[15] 曾翔亮,董希斌,高明.不同诱导改造后大兴安岭蒙古栎低质林土壤养分的灰色关联评价[J].东北林业大学学报,2013,41(7):48-52.
ZENG X L,DONG X B,GAO M.Grey correlation evaluation of soil nutrients to Quercus mongolica low-quality forest in Daxing’an mountains after different induced Transformation[J].Journal of Northeast Forestry University,2013,41(7):48-52.
[16] 唐国华,董希斌,张甜,等.大兴安岭低质林补植改造效果的综合评价[J].东北林业大学学报,2017,45(8):20-24,48.
TANG G H,DONG X B,ZHANG T,et al.Comprehensive evaluation on the effect of replanting alterations of low-quality forest in Daxing′an mountains[J].Journal of Northeast Forestry University,2017,45(8):20-24,48.
[17] 周冬兰,王鑫晨,卢建,等.高强度间伐对马尾松叶片与土壤养分关系的影响[J].森林工程,2024,40(3):11-19.
ZHOU D L,WANG X C,LU J,et al.Effects of thinning on leaf-soil nitrogen and phosphorus relationships in Masson pine plantation[J].Forest Engineering,2024,40(3):11-19.
[18] BOLYEN E,RIDEOUT J R,DILLON M R,et al.Reproducible,interactive,scalable and extensible microbiome data science using QIIME 2[J].Nature Biotechnology,2019,37(8):852-857.
[19] MARTIN M.Cutadapt removes adapter sequences from high-throughput sequencing reads[J].EMBnet,2011,17(1):10.
[20] CALLAHAN B J,MCMURDIE P J,ROSEN M J,et al.DADA2: High-resolution sample inference from Illumina amplicon data[J].Nature Methods,2016,13(7):581-583.
[21] BOKULICH N A,KAEHLER B D,RIDEOUT J R,et al.Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin[J].Microbiome,2018,6(1):90.
[22] KÕLJALG U,NILSSON R H,ABARENKOV K,et al.Towards a unified paradigm for sequence-based identification of fungi[J].Molecular Ecology,2013,22(21):5271-5277.
[23] CHAO A.Nonparametric estimation of the number of classes in a population[J].Scandinavian Journal of Statistics 1984,11(4):265-270.
[24] SHANNON C E.A mathematical theory of communication[J].The Bell System Technical Journal,1948a,27(3):379-423.
[25] SHANNON C E.A mathematical theory of communication[J].The Bell System Technical Journal,1948b,27(3):623-656.
[26] SIMPSON E H.Measurement of diversity[J].Nature,1949,163:688.
[27] PIELOU E C.The measurement of diversity in different types of biological collections[J].Journal of Theoretical Biology,1966,13:131-144.
[28] JACCARD P.Nouvelles recherches sur la distribution florale[J].Bulletin de la Socciete Vaudoise des Sciences Naturelles,1908,44:223-270.
[29] LOZUPONE C,KNIGHT R.UniFrac: a new phylogenetic method for comparing microbial communities[J].Applied and Environmental Microbiology,2005,71(12):8228-8235.
[30] LOZUPONE C A,HAMADY M,KELLEY S T,et al.Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities[J].Applied and Environmental Microbiology,2007,73(5):1576-1585.
[31] ZAURA E,KEIJSER B J F,HUSE S M,et al.Defining the healthy "core microbiome" of oral microbial communities[J].Bmc Microbiology,2009,9(1):259.
[32] BREIMAN L.Random forests[J].Machine Learning,2001,45:5-32.
[33] LIAW A,WIENER M.Classification and regression by random forest[J].R News,2002,2:18-22.
[34] 崔晋瑶.施肥对东北东部典型人工林土壤微生物学特征的影响[D].哈尔滨:东北林业大学,2023.
CUI J Y.Effects of fertilization on soil microbiological characteristics of typical coniferous plantation in Northeast of China[D].Harbin:Northeast Forestry University,2023.
[35] YANG H,PAN C,WU Y,et al.Response of understory plant species richness and tree regeneration to thinning in Pinus tabuliformis plantations in northern China[J].Forest Ecosystems,2023,10(2):215-226.
[36] HAN G,MANNAA M,KIM N,et al.Response of pine rhizosphere microbiota to foliar treatment with resistance-inducing bacteria against pine wilt disease[J].Microorganisms,2021,9(4):688.
[37] ZHANG W,WANG X,LI Y,et al.Pinewood nematode alters the endophytic and rhizospheric microbial communities of Pinus massoniana[J].Microbial Ecology,2021,81(3):807-817.
[38] LI L,TAN J,CHEN F.Bacillus pumilus strain LYMC-3 shows nematicidal activity against Bursaphelenchus xylophilus via the production of a guanidine compound[J].Biocontrol Science and Technology,2018,28(12):1128-1139.
[39] PROENCA D N,SANTO C E,GRASS G,et al.Draft genome sequence of Serratia sp.strain M24T3,Isolated from pinewood disease nematode Bursaphelenchus xylophilus[J].Journal of Bacteriology,2012,194(14): 3764.
[40] MAEHARA N.Reduction of Bursaphelenchus xylophilus (Nematoda : Parasitaphelenchidae) population by inoculating Trichoderma spp.into pine wilt-killed trees[J].Biological Control,2008,44(1):61-66.
[41] WANG H H,WANG Y B,YIN C,et al.In vivo infection of Bursaphelenchus xylophilus by the fungus Esteya vermicola[J].Pest Management Science,2020,76(8):2854-2864.
[42] 杨勇,李昆太.放线菌资源及其活性物质研究概述[J].生物灾害科学,2019,42(1):7-14.
YANG Y,LI K T.The overview of actinomycetes resources and its active substances[J].Biological Disaster Science,2019,42(1):7-14.
[43] OUCHENE R,INTERTAGLIA L,ZAATOUT N,et al.Selective isolation,antimicrobial screening and phylogenetic diversity of marine actinomycetes derived from the Coast of Bejaia City (Algeria),a polluted and microbiologically unexplored environment[J].Journal of Applied Microbiology,2022,132(4):2870-2882.
[44] ZAYA J,DATTA G,MISHRA M,et al.Actinomycetes-the microbial machinery for the organic-cycling,plant growth,and sustainable soil health[J].Biocatalysis and Agricultural Biotechnology,2021,31:101893.
[45] ZHANG D,LU Y,CHEN H,et al.Antifungal peptides produced by actinomycetes and their biological activities against plant diseases[J].Journal of Antibiotics,2020,73(5):265-282.
[46] KIM S K,PARK J E,OH J M,et al.Molecular characterization of four alkaline chitinases from three chitinolytic bacteria isolated from a mudflat[J].International Journal of Molecular Sciences,2021,22(23):12822.
[47] MEHRSHAD M,SALCHER M M,OKAZAKI Y,et al.Hidden in plain sight-highly abundant and diverse planktonic freshwater Chloroflexi[J].Microbiome,2018,6:176.
[48] 张瑛,马雪松,敬如岩,等.基于宏基因组测序技术分析连作对杨树人工林土壤微生物群落的影响[J].山东大学学报(理学版),2019,54(01):36-46.
ZHANG Y,MA X S,JING R Y,et al.Effects of successive-planting poplar plantation on soil microbial community[J].Journal of Shandong University: Natural Science,2019,54(1):36-46.
[49] 龚治翔,马晓寒,任志广,等.连作烤烟根际土壤细菌群落16S rDNA-PCR-DGGE分析[J].中国农业科技导报,2018,20(02):39-47.
GONG Z X,MA X H,REN Z G,et al.Analysis of bacterial communities in rhizosphere soil of continuous cropping flue-cured tobacco using 16S rDNA-PCR-DGGE[J].Journal of Agricultural Science and Technology,2018,20(2):39-47.
[50] HANADA S.Filamentous anoxygenic phototrophs in hot springs[J].Microbes and Environments,2003,18(2):51-61.
[51] SOROKIN D Y,VEJMELKOVA D,LÜECKER S,et al.Nitrolancea hollandica gen.nov.,sp nov.,a chemolithoautotrophic nitrite-oxidizing bacterium isolated from a bioreactor belonging to the phylum Chloroflexi[J].International Journal of Systematic and Evolutionary Microbiology,2014,64(6):1859-1865.
[52] SOROKIN D Y,LÜECKER S,VEJMELKOVA D,et al.Nitrification expanded: discovery,physiology and genomics of a nitrite-oxidizing bacterium from the phylum Chloroflexi[J].Isme Journal,2012,6(12):2245-2256.
[53] ZARZYCKI J,BRECHT V,MÜELLER M,et al.Identifying the missing steps of the autotrophic 3-hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus[J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(50):21317-21322.
[54] CLINE L C,ZAK D R.Soil microbial communities are shaped by plant-driven changes in resource availability during secondary succession[J].Ecology,2015,96(12):3374-3385.
[55] LIU J,SUI Y,YU Z,et al.Diversity and distribution patterns of acidobacterial communities in the black soil zone of northeast China[J].Soil Biology & Biochemistry,2016,95:212-22.
[56] 高明,朱玉杰,董希斌.采伐强度对大兴安岭用材林土壤化学性质的影响[J].东北林业大学学报,2013,41(12):39-41,76.
GAO M,ZHU Y J,DONG X B.Effects of tending felling on soil chemical property of timber forest in Daxing’an Mountain[J].Journal of Northeast Forestry University,2013,41(12):39-41,76.
