
基于H&E图像和基因表达数据的多模态深度学习模型预测胃癌生存风险
2024-08-06 00:00:00马艳雨贺平安
软件工程 2024年8期




关键词:胃癌;H&E染色图像;基因表达;深度学习;多模态
中图分类号:TP391.41 文献标志码:A
0 引言(Introduction)
癌症是导致患者死亡的主要原因,其中胃癌是常见的消化系统恶性肿瘤之一,其发病率和死亡率近年来居高不下,严重威胁人类健康[1]。准确预测胃癌患者的死亡风险对于临床决策、治疗计划和患者心理调整至关重要。病理学家一般通过苏木精和伊红(Hematoxylin-Eosinstaining,H&E)染色的图像获得肿瘤形态和组织学信息。数字成像技术的进步使得全幻灯片扫描仪在病理学图像数字化中得到广泛应用。深度卷积神经网络(CNN)已成为一种重要的图像分析工具,数字成像技术和CNN结合应用,极大地提高了病理学领域的专家、学者及研究人员的工作效率,为肿瘤诊断和治疗提供了更准确的依据[2-4]。
转录组学分析关注mRNA作为DNA和蛋白质之间的中间分子,其表达水平和表达模式与肿瘤发展、细胞功能及患者预后密切相关[5]。综合考虑组织病理学和基因组学的互补信息可以提高生存风险预测的潜力。然而,以往的研究多专注于各自领域的个体模式,缺乏对信息的整合,限制了对癌症生物学的全面理解[6]。
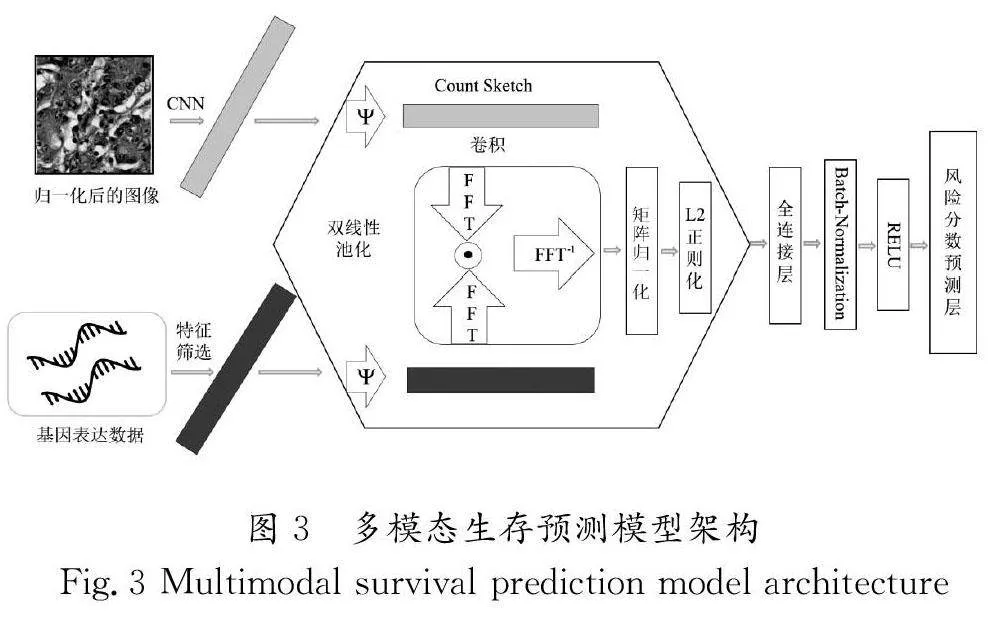
因此,本研究通过整合H&E染色图像的特征和基因组数据,构建了一个多模态生存预测模型,以提高癌症患者生存预测的准确性。具体而言,首先,利用ResNet18从H&E染色区域提取深层特征;其次,采用多模态紧凑双线性池化(MCB)方法融合病理学和基因组学的特征;最后,将融合后的多模态特征输入Cox回归层,得到癌症患者的风险分数,为个体化的治疗和关怀提供有力支持。
1 相关理论(Related theory)
1.1 H&E染色图像
H&E染色图像是常见的组织病理学图像,通过苏木精和伊红染色,使得细胞核和细胞质分别呈现蓝色和粉红色,从而提供了关于组织结构和细胞形态的重要信息。制备过程包括组织固定、脱水、清理、染色、包埋、切片、脱蜡、染色和封片等步骤。H&E染色图像的高分辨率导致每张图像的像素值达到10亿左右,对图像处理带来了巨大的挑战。像素数量庞大可能会引起计算的问题,因此通常需要将图像切割成小块以提高处理效率。此外,图像在不同设备和条件下可能存在颜色偏差,会影响可比性和分析的准确性。因此,采用颜色归一化处理操作是必要的,它可以确保图像的一致性,并提升建模和分析的可靠性。
1.2 生存风险预测问题
生存风险预测问题通常被视为一个重要的临床挑战,其中的关键在于预测患者在特定时间点(例如3年)的生存状态,并将其二分化为生存或死亡[7]。然而,使用传统的分类方法建立模型存在一些局限性,尤其在随访数据不完整的情况下更为明显。在这种情况下,时间-事件模型,如Cox回归[8]和随机生存森林[9]展现出了更为灵活的能力。
与传统分类方法不同,时间-事件模型可以利用所有受试者的信息进行训练,并在各种时间点预测患者的生存概率。这一方法不仅更全面地考量了患者的生存状况,还能有效应对随访数据缺失的问题,建模过程更稳健和可靠。医生和研究人员借助时间-事件模型,可以更精准地评估患者的生存风险,并制定个性化的治疗方案和临床管理策略。
2 数据与方法(Data and method)
2.1 数据预处理
2.1.1 数据集下载及预处理
本文使用的H&E染色图像和转录组数据均来自https:∥gdc.cancer.gov/的癌症基因组图谱(TCGA)。通过患者识别号(TCGA ID)进行数据整合和匹配,最终得到320例胃癌(STAD)患者的H&E图像数据和基因表达数据。
2.1.2 H&E图像的预处理
为了从H&E图像中提取特征,本研究采取了一系列预处理操作,H&E图像预处理步骤如图1所示。首先,由一位经验丰富的病理学家对肿瘤区域进行了视觉注释,确保分析区域的正确性。其次,针对像素极大的H&E染色图像,为了得到适配模型的大小,对H&E图像进行切割,形成512×512的小图像块,针对切割后的图像,计算出肿瘤区域与总区域的比率,并舍弃了比率小于0.3的存在噪声的小图块(如空白或模糊的区域)。最后,使用文献[10]中提到的颜色归一化算法对带有有用信息的小切片进行颜色归一化,消除不同H&E图像的染色差异[10]。
2.1.3 基因表达数据的预处理
为了减少基因表达数据的冗余信息,首先,删除在少于50个样本中表达的基因,此操作将胃癌的基因表达数据减少至19 531维。其次,利用单因素Cox回归对与生存相关的特征进行筛选,并设定P 值小于0.05为阈值。在此过程中,共筛选出1 385个与生存相关的基因。
2.2 基于H&E染色图像的深度学习模型建立
为了解决深度神经网络训练过程中的一系列问题,HE等[11]提出了残差网络(ResNet),这一结构由一系列残差块组成。每个残差块经过多次卷积操作后,将其输出与输入相加。通过引入残差连接,ResNet允许某一层的输出直接跳过一个或多个层,连接到后续层的输入。这种结构使得网络能够更有效地学习复杂的特征,同时缓解了深度网络训练过程中的退化、梯度消失和梯度爆炸等问题。
ResNet18作为一种经典的深度卷积神经网络,其构架由17个卷积层和1个全连接层组成。该网络的结构可以划分为4个主要模块,从第一层开始,每个模块包含两个基本块,基本块由一对卷积层组成,每个卷积层后面跟随着ReLU激活函数和批量归一化层(Batch Normalization),以增强模型的非线性建模能力和稳定性。在整个ResNet18中,这些基本块的堆叠形成一个强大的特征提取和学习结构,使得网络能够有效地捕获和表示输入数据的复杂特征。此外,每个基本块中的卷积层都设计有残差连接,以促进梯度的顺畅传播和模型的训练收敛。最终的全连接层将卷积层提取的高层次特征映射为一维向量。深度生存预测模型架构如图2所示。
2.3 基于多模态深度学习的生存模型的建立
本研究采用基于多模态紧凑型双线性池化(MCB)的多模态融合算法[12],旨在将H&E图像的特征与基因表达特征融合为一个新的特征向量。相对于传统的向量融合方法(如拼接、逐位相乘、逐位相加等),MCB采用了一种端到端学习的反向传播方法,在处理双线性特征时,MCB通过多项式核的内积近似降低特征维度,确保模型性能损失最小。
双线性池化是通过计算特征向量的外积实现融合,充分利用了向量元素之间的交互作用。该过程包括对每个位置的特征向量进行向量外积计算,将所有位置的计算结果进行求和池化,最终得到特征向量。为了应对高维特征的挑战,采用了近似多项式核的算法(Tensor Sketching)进行降维,即紧凑型双线性池化(CBP)。
在多模态情境下,MCB对CBP中的近似多项式核的算法进行改进。具体工作流程包括提取各模态对应的特征,通过预训练的CNN提取图像高层特征,采用多层感知机(MLP)对其他模态进行特征提取;利用Count Sketch方法对两个模态的特征进行逼近,得到降维后的特征;在频域内进行向量内积计算,通过FFT-1 逆变换至时域空间,最后对得到的时域特征进行矩阵归一化和L2正则化。
多模态生存预测模型架构如图3所示,首先将归一化后的病理图像块输入ResNet18网络,在全连接层得到512维的特征向量。同时,采用MLP处理经预处理后的基因表达数据,得到相应的特征向量。其次利用MCB方法将H&E图像特征和基因表达特征的特征进行融合,并将融合后的特征输入生存风险预测层,进行端到端训练。这一整合方法旨在综合利用不同模态的信息,提高生存风险预测模型的预测准确性。
3 实验结果与分析(Experimental results andanalysis)
3.1 数据集划分与模型训练
本研究按照五折交叉验证的方式将样本划分为训练集和测试集,确保每折在训练集和测试集中存活样本和死亡样本的比例相同,其数据划分与模型训练示意图如图4所示。
在训练数据方面,采用数据扩充和标准化策略。具体而言,通过随机水平翻转和面片的随机仿射变换(保持中心不变)进行数据增强。在RGB通道上进行z-score归一化处理后,将增强的小图像块进行中心裁剪(像素为224×224),确保训练数据的高质量和一致性。对于测试数据,仅进行标准化处理,在确保数据高质量和一致性的同时避免引发不必要的变化。
在模型选择方面,使用随机梯度下降(SGD)算法更新模型的参数,将学习率设置为2×10-3。所有模型至少训练20个epochs,最多可达300个epochs。在训练过程中监控模型的性能,若在第50个epochs之后,训练损失连续10个epochs未减少,则提前停止。最低损失的最佳模型将用于测试模型性能。
在风险分数的预测阶段,针对每位患者,取其所有小图像块预测出的风险分数的平均值作为该患者的风险分数。
3.2 评价指标
本研究采用c-index,即一致性指数评价模型的预测能力。c-index的计算方式如下。
(1)把所有研究对象随机地两两组成对子。
(2)对于每一对患者,若生存时间较长的样本,其预测的生存时间也较长,预测生存概率较高的样本,其实际生存概率也较高,则表示预测结果与实际结果一致。
(3)计算c-index=K/M,其中:K是一致的对子数,M 是有用的对子数。
c-index的值为0.5~1,0.5表示预测结果与实际结果完全不一致,说明该模型没有预测作用;1表示预测结果与实际结果完全一致,说明该模型具有预测作用。
3.3 模型的性能比较
3.3.1 不同模态的性能比较
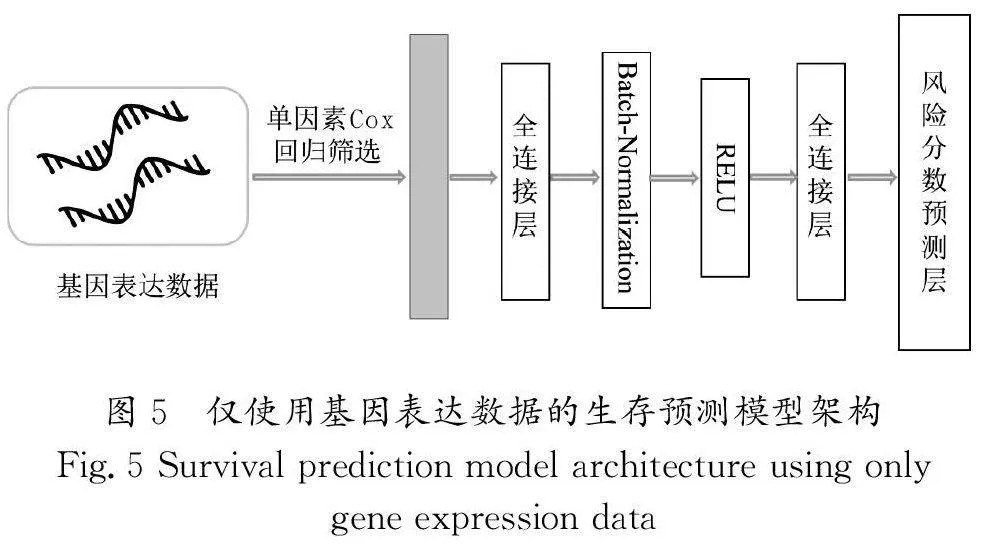
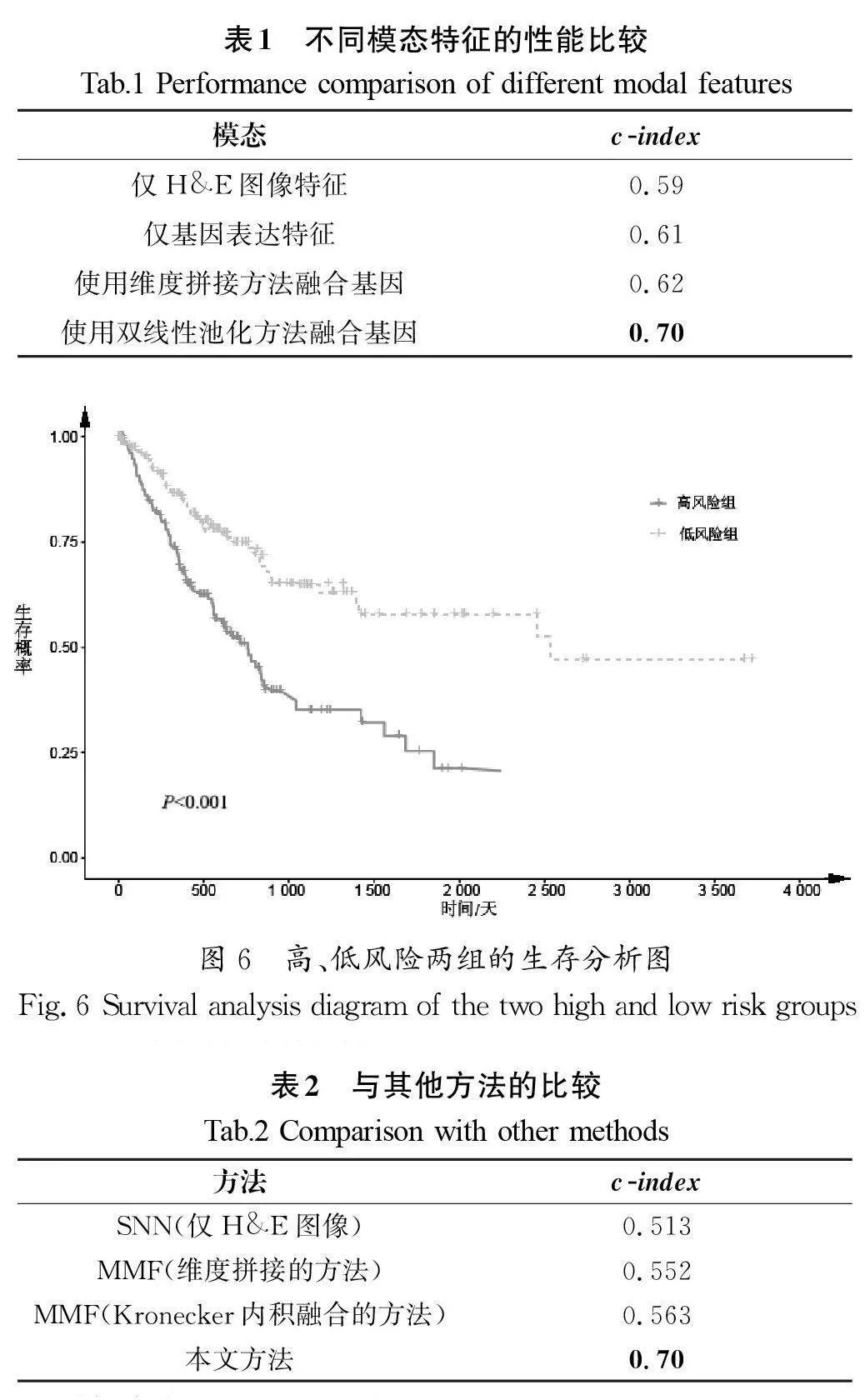
为了评估多模态方法的有效性,本研究使用MCB方法的模型与仅使用H&E图像数据的模型以及仅使用基因表达数据的模型进行了比较。针对仅使用基因表达数据的建模流程,使用单因素Cox回归筛选出的1385维基因表达特征作为输入,保持全连接层结构不变,用于预测胃癌患者的风险分数,仅使用基因表达数据的生存预测模型架构如图5所示。研究人员对各模态特征的预测能力进行了对比,结果如表1所示。
通过对比表1中的结果发现,使用MCB融合方法的模型性能明显优于仅使用单一模态特征和使用维度拼接方法的模型性能,表明使用双线性池化方法融合基因的方法在胃癌生存风险预测中具有显著的优势。
在测试集上,将模型预测出的风险分数以中位数为阈值,将样本分为两组,即高风险组和低风险组,并对这两组样本进行生存分析。为了检查两组样本之间的差异,使用了log-rank 进行检验,生存分析的结果如图6所示,两组样本之间的P 值远小于0.05。以上结果表明,该模型能够有效地将胃癌患者分成两组,并且这两组之间存在显著差异。
3.3.2 与不同方法的比较
为了评估本研究提出的模型的性能,将其与文献[4]中提到的所有方法进行比较,具体结果如表2所示。表2中结果表明,本研究提出的方法明显优于其他方法。
4 结论(Conclusion)
本研究采用了基于MCB的多模态融合算法,成功地将H&E图像的特征与基因表达特征融合为一个新的特征向量,用于预测胃癌患者的生存风险。与仅使用H&E染色图像或采用维度拼接的方法相比,MCB算法显著提高了模型性能。这一多模态融合方法为胃癌患者生存风险预测提供了更全面的信息。传统的单一模态方法可能无法充分捕捉到患者病理特征与基因表达之间的复杂关系,采用MCB算法能够将不同模态的信息有机地结合起来,从而获得更准确、更全面的特征向量,进而提高了预测模型的性能。
此外,通过对高、低风险两组患者的生存情况进行分析发现,该多模态算法模型能够有效地将胃癌患者分层。这一发现不仅对个体患者的治疗和管理提供了重要的指导,也为临床实践提供了有力支持。
作者简介:
马艳雨(1998-),女,硕士生。研究领域:人工智能,生物信息学。
贺平安(1969-),男,博士,教授。研究领域:计算生物学,生物信息学。
