
卟啉化合物芳香性的理论研究
2024-05-10 01:20:50周雯欣赵文伟
山东化工 2024年6期
周雯欣,赵文伟
(临沂大学 化学化工学院,山东 临沂 276005)
卟啉类化合物是由次甲基链接四个吡咯环组成的一个大环共轭化合物,这类化合物大环具有18π电子的共轭路径,具有芳香性。除此之外,两个吡咯环分别具有6个π电子,根据休克尔规则,也具有芳香性[1]。其丰富的π电子共轭体系,一直是芳香性的研究的模型化合物。
Dideazaporphyrin与无取代基卟啉(卟吩)相比,少了两个NH基,如图1所示,dideazaporphyrin只有18π电子的芳香性[2]。而卟吩除了18π电子的芳香性外,还有两个吡咯环具有芳香性。但是,卟吩的1H核磁共振光谱与dideazaporphyrin的1H核磁共振光谱十分相似,所以卟吩的芳香被认为“be encapsulated in the diaza[18]annulene substructure”[2],卟吩的芳香性与dideazaporphyrin的芳香性类似。这些研究表明卟啉分子芳香性的主要来源于18π电子的大环共轭路径,卟啉分子中吡咯环的贡献很小。
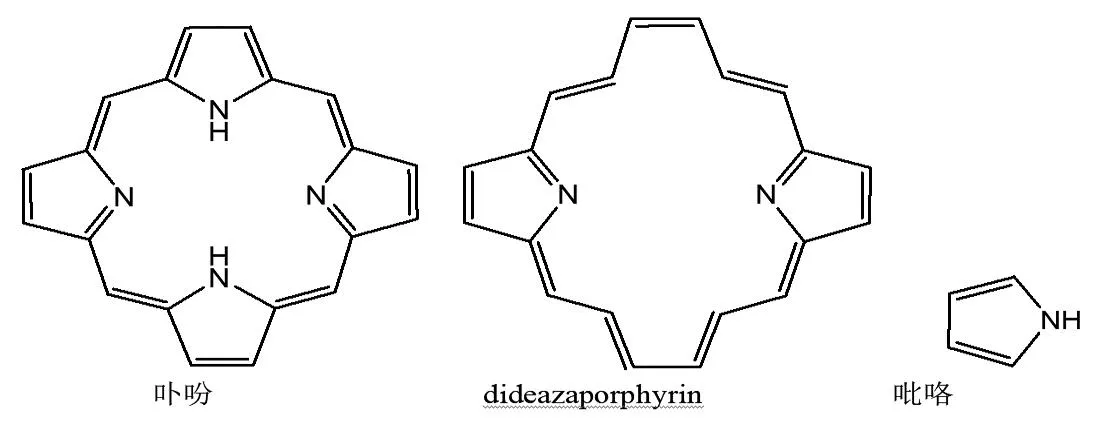
图1 卟吩、dideazaporphyrin和吡咯分子结构
卟啉化合物中两个吡咯环对整个卟啉分子芳香性的贡献一直存在争论。Cyranski等人使用NICS(核独立化学位移)计算研究卟吩的芳香性,认为吡咯环上的NH基是芳香性的一部分,而且18π电子16轮烯的内交叉共轭路径比较重要[3]。但是Fliegl等人基于诱导环电流的计算表明NH基有很大诱导电流阻力[4]。Wu等人使用BLW(Block-localized wave lunction)方法研究表明卟啉的芳香性稳定化能来源于大环和吡咯环的贡献,并且吡咯环的贡献更有效[1]。因此有必要对卟啉化合物的芳香性开展进一步的理论研究。
随着量子化学计算方法的发展,有很多的理论方法可以用来研究分子的芳香性。诱导环电流密度的各向异性(Anisotropy of Induced Current Density,AICD)是通过模拟分子中电子的感应电流来判断芳香性的方法[5]。芳香性分子由于π电子的离域,芳香性分子能量会比非芳香性分子的能量低,芳香稳定化能(Aromatic stabilization energy,ASE)通过等键化学反应计算芳香分子的能量降低的大小判断分子的芳香性。AdNDP(Adaptive natural density partitioning,适应性自然密度划分)方法是将分子的电子密度最大可能划分为定域形式的电子对,也就是单中心2电子键、两中心2电子键、三中心2电子键等[6]。如果电子密度不能完全定域某些原子之间,它就具有芳香性,如果苯分子就具有三个6中心2电子键,这些键是离域在6个碳原子上的。
对无取代基卟啉(卟吩)、dideazaporphyrin和吡咯等化合物进行理论研究。计算以上化合物的诱导环电流密度的各向异性、芳香性稳定化能和AdNDP电子对,研究以上化合物的芳香性,为卟啉类化合物的芳香性提供一个合理的解释。解释卟啉类化合物的1H NMR 为什么跟dideazaporphyrin的1H NMR十分相似,吡咯环的芳香性对卟啉分子芳香性贡献等问题。相比于实验对芳香性的研究只能借助于核磁共振光谱,理论计算可以从芳香化合物的诱导环电流密度、稳定性以及化学键特征等方面对芳香性进行研究,提供关于卟啉芳香性的更多的信息。
1 计算方法
采用B3LYP/6-31+G(d,p)方法对卟吩、dideazaporphyrin和吡咯等化合物进行几何优化,得到稳定的几何结构。计算使用Gaussian 09程序完成。为了能对卟啉类化合物的芳香性有全面的了解,对芳香性的使用多种理论方法进行研究,包括AICD法,计算芳香稳定化能和AdNDP法。诱导环电流密度的各向异性(AICD)使用AICD2.0 程序计算。芳香稳定化能通过构建一个等键的化学反应进行计算,要求反应前后的C=C双键数量相等,和反应前后sp2杂化C原子与sp2杂化C原子间的化学键数量相等。对同一个芳香性化合物,可以构建出不同的等键反应,计算得到的芳香性稳定化能也存在一定的差别[7]。等键化学反应前后的化学物质均使用B3LYP/6-31+G(d,p)方法优化并计算能量。AdNDP(适应性自然密度划分)分析使用Multiwfn 3.8程序完成。
2 结果与讨论
2.1 诱导环电流密度的各向异性(AICD)分析
吡咯、dideazaporphyrin和卟吩的π电子AICD的等值面(isovalue=0.06 au)如图2所示。吡咯单体分子和dideazaporphyrin分子都有诱导环电流,表明这两个分子都具有芳香性。当等值面的电流密度为0.065 au时,吡咯单体分子与NH基相对的β位C原子之间的π电子的诱导环电流开始断开,而dideazaporphyrin和卟吩的π电子诱导换电流依旧很强。说明吡咯单体分子的6π电子诱导环电流比dideazaporphyrin的18π电子诱导环电流要弱。
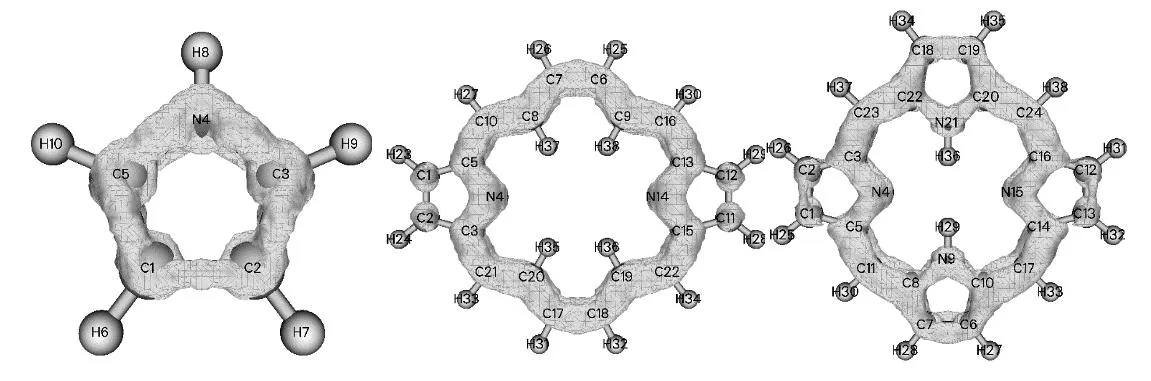
图2 吡咯、dideazaporphyrin和卟吩π电子诱导环电流密度的各向异性等值面图(isovalue=0.06 au )
如图2所示,卟吩的π电子诱导环电流在吡咯环的两个β位C原子之间和NH基上都有分布,但是NH基诱导环电流密度要比β位C=C原子间的π电子诱导环电流密度要弱,与之前的研究结果一致[4]。如果不考虑NH基上π电子诱导环电流,dideazaporphyrin的诱导环电流和卟吩的π电子诱导环电流密度高度相似,进而感应磁场也就是H NMR谱也十分相似,这也是为什么卟啉的芳香性被认为与dideazaporphyrin芳香性类似的原因。
2.2 芳香稳定化能分析
芳香性化合物比没有芳香性的物质要稳定,芳香稳定化能(Aromatic stabilization energy,ASE)是通过比较芳香性物质的能量和非芳香物质能量研究分子芳香性的一种方法,表明π电子的离域化导致的芳香性分子体系能量的降低[7]。芳香稳定化能通过等键的化学反应进行计算,不同的等键化学反应,计算得到的芳香稳定化能也可能不同。等键化学反应要求反应物和产物具有相同数量的单键和双键,相同杂化原子间的化学键数量也相等。卟吩、dideazaporphyrin和吡咯等化合物芳香稳定化能的计算结果列在表1中。

表1 三种化合物的芳香性稳定化能
根据文献报道,吡咯的芳香稳定化能使用图3的等键化学反应计算[7],在B3LYP/6-31+G(d,p)水平上计算得到的吡咯的芳香稳定化能是19.24 kcal·mol-1。Dideazaporphyrin的芳香稳定化能使用图4的等键化学反应计算,在B3LYP/6-31+G(d,p)水平上计算得到的Dideazaporphyrin的芳香稳定化能是14.76 kcal·mol-1。这表明吡咯的芳香稳定化能比Dideazaporphyrin芳香稳定化能要高。这个结论与AICD 诱导环电流密度的结论相反,Dideazaporphyrin的π电子诱导环电流密度比吡咯的π电子诱导环电流密度要大。
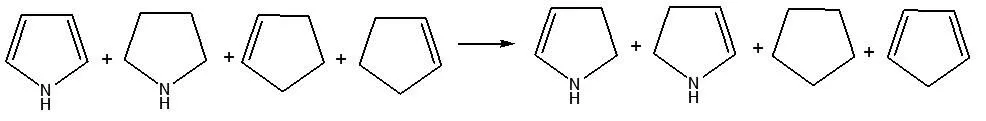
图3 计算吡咯分子芳香稳定化能的等键反应
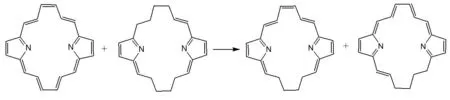
图4 计算dideazaporphyrin分子芳香稳定化能的等键反应
卟吩的分子结构远比Dideazaporphyrin和吡咯复杂,我们根据吡咯芳香性稳定化能的计算方法,构建了等键的化学反应用于计算卟吩的芳香稳定化能,如图5所示。图5所示的等键化学反应在B3LYP/6-31+G(d,p)水平计算得到的芳香稳定化能是50.72 kcal·mol-1。
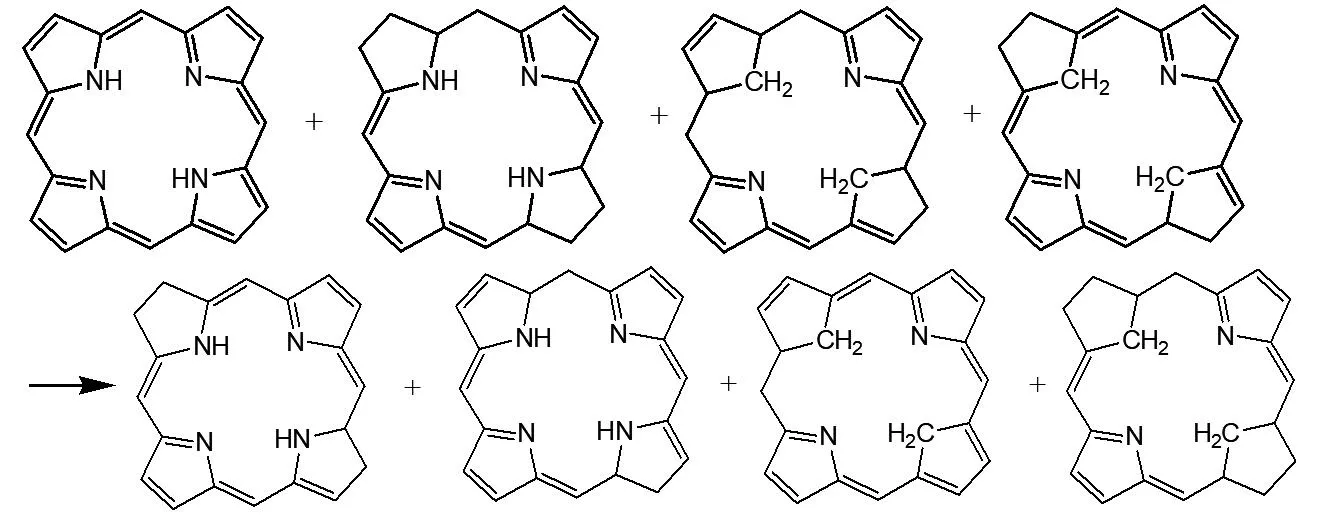
图5 计算卟吩分子芳香稳定化能的等键化学反应
从分子结构上看,卟吩的芳香性由18π电子的大环芳香性和两个6π电子的吡咯环芳香性组成,那么卟吩分子的芳香稳定化能与dideazaporphyrin和两个吡咯芳香稳定化能的和相近。Dideazaporphyrin和两个吡咯芳香稳定化能的和是53.24 kcal·mol-1,和图5的等键化学反应计算的卟吩的芳香稳定化能(50.72 kcal·mol-1)十分接近。两个吡咯分子的芳香稳定化能之和是38.48 kcal·mol-1,远大于Dideazaporphyrin 的芳香稳定化能14.76 kcal mol-1,与BLW方法的研究结果一致[1]。这说明在卟吩分子中两个吡咯对芳香性稳定化能的贡献十分重要,吡咯环的芳香性应该是卟吩芳香性的一部分。
2.3 适应自然密度划分(AdNDP)分析
AdNDP方法将分子的电子密度尽可能地划分为定域化的形式,如1中心2电子键(孤对电子),2中心2电子键,3中心2电子键。芳香性分子π电子是离域的,有多个多中心2电子化学键[6]。使用AdNDP方法对吡咯、Dideazaporphyrin和卟吩分子进行了分析。AdNDP分析产生1中心2电子键对应孤对电子,大部分2中心2电子键对应σ键,这些电子对对分析分子的芳香性没有作用,所以不讨论这些AdNDP分析结果。
吡咯是6 π电子的共轭体系,根据休克尔规则,具有芳香性。AdNDP分析结果如图6所示,吡咯分子具有三个5中心2电子的π键,并且这三个5中心2电子键是离域的,在四个碳原子核一个氮原子上都有分布,因此,吡咯具有芳香性。
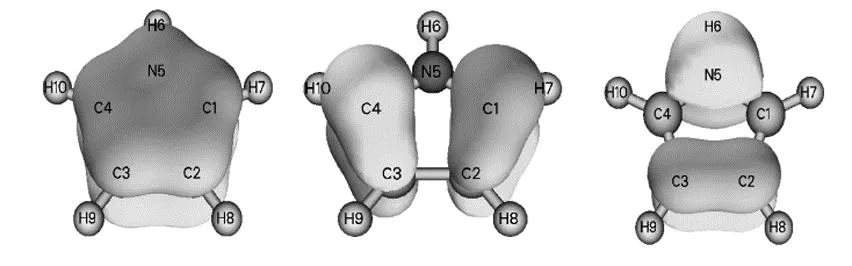
图6 吡咯分子的AdNDP分析结果(三个5中心2电子π键)
对Dideazaporphyrin分子的AdNDP分析如图7所示。在每一个无氢吡咯环上都有一个2中心-2电子的π键,这种π键处在无氢吡咯环β位C=C键上,表明这两个C=C键上的π电子没有参与18π电子的大环共轭体系中,与AICD的分析结果一致。除此之外,还有六个3中心-2电子π键和三个18中心-2电子π键。六个3中心2电子π键其中有两个分布在无氢吡咯环的C-N-C基团上,这样每个无氢吡咯环上各有一个2中心-2电子的π键和一个3中心-2电子的π键。Dideazaporphyrin分子的三个18中心-2电子的π键是完全离域的,分布在18个原子上,表明Dideazaporphyrin分子是具有芳香性的。
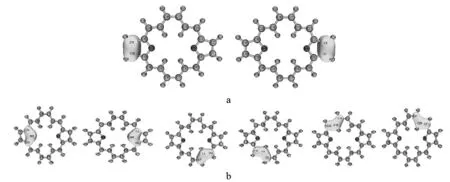
(a)两个2中心2电子π键;(b)六个3中心2电子π键;(c)三个18中心2电子π键。图7 Dideazaporphyrin分子的AdNDP分析结果
卟吩分子的AdNDP分析结果如图8所示,在每个无氢吡咯环上,各有一个2中心-2电子键和一个3中心2电子π键,与Dideazaporphyrin分子的无氢吡咯环的AdNDP分析结果相同。在每个有氢吡咯环上,有两个5中心-2电子π键,这两个5中心-2电子π键特征与吡咯单体分子的两个5中心-2电子π键完全一致。与吡咯单体分子相比,卟吩分子中的有氢吡咯环缺少了一个5中心-2电子π键,这部分π电子将参与形成18中心-2电子π键。这些AdNDP的结果表明,在卟吩分子中,吡咯作为分子一部分,仍具有芳香性。除此之外,卟吩分子还有五个18中心-2电子π键。这些π键是离域的,分布在18个原子上,表明卟吩分子除了吡咯环具有芳香性外,大环还具18π电子的芳香性。也表明卟吩的芳香性要比Dideazaporphyrin分子的芳香性要复杂。不能认为卟吩的芳香性与dideazaporphyrin芳香类似,因为卟吩的吡咯环还具有芳香性。
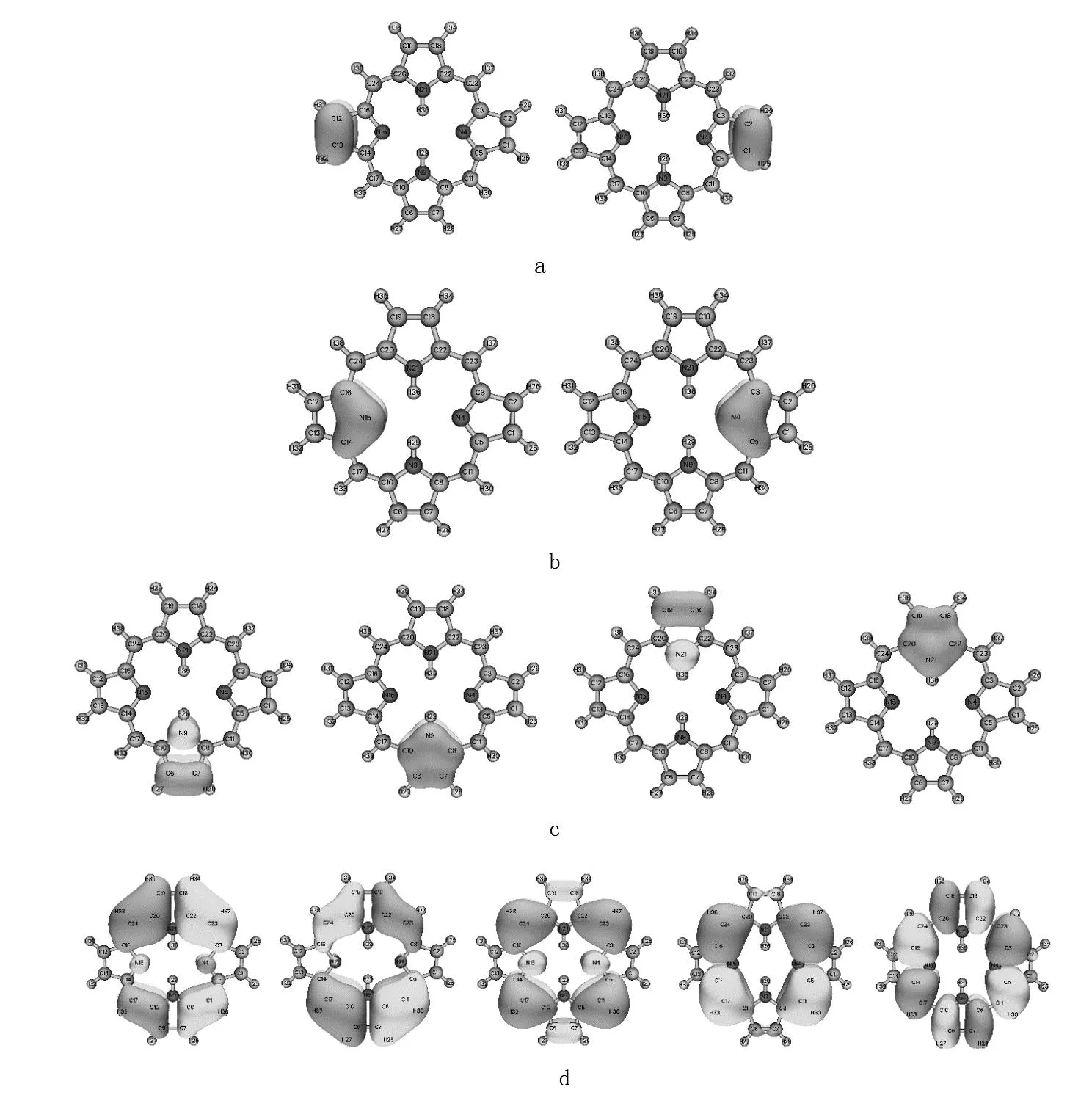
(a)两个2中心2电子π键;(b)两个3中心2电子π键;(c)四个5中心2电子π键;(d)五个18中心2电子π键。图8 卟吩分子的AdNDP分析结果
3 结论
通过诱导环电流密度的各向异性图、芳香性稳定化能和AdNDP法等多种理论研究方法分别对卟吩、dideazaporphyrin和吡咯分子的芳香性进行了理论研究。从分子结构上看,dideazaporphyrin比卟吩分子少了两个NH基,只有一个18π电子的共轭路径。而卟吩分子除了具有18π电子的共轭路径外,还有两个吡咯环具有6π电子的芳香性。
对卟吩、dideazaporphyrin和吡咯分子绘制了π电子诱导环电流密度的等值面图。吡咯的6π电子诱导环电流密度要比dideazaporphyrin的18π电子诱导环电流密度要小。这表明在卟吩分子中,18π电子的大环诱导环电流密度要比吡咯环的6π诱导环电流密度要大,当18π电子的大环诱导环电流和6π诱导环电流相反时,吡咯环的6π环电流就会被18π电子的大环诱导环电流抵消,在卟吩分子中只有18π电子的大环诱导环电流出现。因此,卟吩和dideazaporphyrin π电子诱导环电流密度的等值面图十分相似。实验上卟吩和dideazaporphyrin分子的H NMR谱也十分相似。这也就是为什么卟吩的芳香性被认为是dideazaporphyrin芳香性的原因。
通过构建等键化学反应计算了卟吩、dideazaporphyrin和吡咯分子的芳香稳定化能。在B3LYP/6-31+G(d,p)水平上计算得到的吡咯的芳香稳定化能是19.24 kcal·mol-1。而Dideazaporphyrin分子的芳香稳定化能是14.76 kcal·mol-1。Dideazaporphyrin的芳香稳定化能比吡咯的芳香稳定化能要低4.48 kcal·mol-1,与AICD 诱导环电流密度的结论相反。卟吩分子结构比较复杂,我们构建了等键的化学反应,在B3LYP/6-31+G(d,p)水平计算得到的芳香稳定化能是50.72 kcal·mol-1。卟吩的芳香性由18π电子的大环芳香性和两个6π电子的吡咯环芳香性组成,卟吩分子的芳香稳定化能应该是18π电子大环芳香稳定化能和两个吡咯环芳香稳定化能之和。Dideazaporphyrin和两个吡咯芳香稳定化能的和是53.24 kcal·mol-1,等键化学反应2计算的卟吩的芳香稳定化能(50.72 kcal·mol-1)十分接近。这说明在卟吩分子中两个吡咯对芳香性稳定化能的贡献十分重要,吡咯环的芳香性应该是卟吩芳香性的一部分。
AdNDP分析表明吡咯单体分子具有三个5中心-2电子的π键,这三个轨道是离域的,吡咯分子具有芳香性。Dideazaporphyrin分子有三个18中心-2电子的π键,也具有芳香性。卟吩分子的每个吡咯环上有两个5中心-2电子的π键,大环上还有五个18中心-2电子π键。这表明卟吩分子中除了吡咯环具有芳香性外,大环还具有芳香性。因此可以推断卟吩的芳香性由大环的芳香性和吡咯环芳香性共同组成,要比Dideazaporphyrin分子以及18轮烯的芳香性要复杂。
猜你喜欢
大学化学(2021年8期)2021-09-26 10:50:46
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30 06:36:44
山东化工(2020年5期)2020-04-07 09:59:30
少儿美术(2019年8期)2019-12-14 08:06:58
少儿美术(快乐历史地理)(2018年7期)2018-04-02 19:58:31
岭南音乐(2017年3期)2017-07-18 11:59:40
合成化学(2015年10期)2016-01-17 08:56:06
传奇故事(破茧成蝶)(2015年1期)2015-02-28 09:26:48
厦门大学学报(自然科学版)(2014年2期)2014-08-06 11:26:48
华东师范大学学报(自然科学版)(2014年4期)2014-03-11 16:18:28
