
大气氧化能力量化研究
2024-05-06 06:50王跃思刘子锐胡波王润玉
大气科学 2024年1期
王跃思 刘子锐 胡波 王润玉 , 2
1 中国科学院大气物理研究所, 北京 100029
2 中国科学院大学, 北京 100049
1 引言
大气是一个复杂的物理与化学体系,也是一个开放的动态系统,与海洋、陆地和生态圈层之间密切相互作用。各圈层向大气中排放的活性微量气体,往往是通过大气氧化反应被清除脱离大气,清除速率与大气的氧化能力有关,表观为大气氧化性的强弱。大气氧化能力(Atmospheric Oxidation Capacity,AOC)的传统定义是大气通过氧化过程去除其中活性微量气体成分的速率总和(Prinn, 2003)。在对流层和近地层大气中,大气氧化能力主要表观为对污染气体的清除能力或净化能力(Cheng et al.,2007; 林云萍和赵春生, 2009),亦称大气氧化性。地球大气形成初期是一个还原性体系,如果没有大气氧化过程,大气的化学成分及天气过程就会和我们今天所见显著不同。
羟基(OH, hydroxyl radical)自由基是大气中最活跃的氧化剂,尽管其在大气中的浓度非常低,但有很高的化学活性,能与大气中绝大多数痕量组分发生化学反应(Seinfeld and Pandis, 2016)。参与对流层大气化学过程的主要氧化剂除OH 自由基外,还有过氧自由基(HO2和RO2, hydroperoxyl and organic peroxyl radicals)、硝基自由基(NO3,nitrate radical)和卤素自 由基等(Thornton et al.,2010)。经自由基引发形成的O3、HONO、HCHO、H2O2、N2O5、HNO3和PAN 等分子型次生氧化剂,被称之为自由基的“储库分子”。白天光照情况下,储库分子可光解再次产生OH,使自由基反应链加长;夜间NO3自由基在气相氧化反应中最为重要,而分子型氧化剂(O3、H2O2、N2O5和HNO3等)参与液相和固相非均相化学反应亦十分活跃(葛茂发等, 2009)。包含在大气液滴和颗粒物中的分子型氧化剂对大气污染物的形成和清除作用也不可小觑,这类非均相化学过程在高湿度、高污染条件下往往会成为氧化反应的主导,但以往研究并未将其计入AOC 的一部分。本文将对这一问题进行重点探讨。
大气氧化能力对大气二次颗粒物和臭氧污染形成具有关键作用,可以说是两种表象,一个本质,然而国内外对其量化研究却十分有限。经历了半个多世纪,美欧等发达国家或地区尚未完全解决大气臭氧污染问题,亦表现出对AOC 的认知仍显不足。虽然早有经典文献报道,可遵循EKMA 曲线通过调控前体物氮氧化物(NOx, nitrogen oxides)和挥发 性 有 机 物(VOCs, volatile organic compounds)控制臭氧产率(Kelly and Gunst, 1990),但由于前体物(尤其是VOCs)的种类和化学活性及其气象环境条件的巨大差异(Wang et al., 2021),使得这一方法很难付诸实施,更无法揭示我国大气氧化能力对二次颗粒物和臭氧污染形成的准确定量作用。国际上关于AOC 的研究大多集中在氧化剂与人为/自然源VOCs 光化学产生O3和二次有机气溶胶(SOA, secondary organic aerosol)的机制、潜势以及环境效应研究等方面,研究手段主要是运用烟雾箱结合数值模式,模拟大气环境下OH、Cl 和O3等氧化剂与各种VOCs 相互作用的动力学和热力学过程,为模式提供参数化方案(Surratt et al., 2010;Carlton et al., 2010; Zhang et al., 2015; Wang et al.,2020)。研究呈现出三种发展趋势:(1)由单一均相气相过程向更加复杂的气—固、气—液和液—固等非均相过程转变;(2)由单一VOCs 物种研究向多物种、实际大气VOCs 复合研究转变;(3)由自然源VOCs 向人为源或混合源VOCs 研究转变。以期获得更加接近真实大气情况下各种氧化过程的认识(Laskin et al., 2015)。
我国最初对AOC 的研究主要集中在光化学污染最重要的标志物——O3。20 世纪70 年代,北京大学唐孝炎首先发现我国兰州西固地区O3污染严重,直到80 年代,该地区O3浓度还经常超过400 μg/m3(Tang et al., 1989)。90 年代开始,北京相继出现高浓度O3污染,1997 年夏季,臭氧峰值平均一度达到240 μg/m3(张远航等, 1998),而文献记载最严重一次臭氧污染发生在2005 年夏季的北京昌平,小时均值高达615 μg/m3(Wang et al.,2006)。当城市O3污染引起关注后,我国近地面O3污染研究相继在多地展开,但主要集中在污染最为严重的华北、长三角、珠三角和成渝等重点城市群区域(Xue et al., 2021),研究方法主要是外场观测和数值模式相结合,研究区域主要集中在近地面与人群直接接触的环境大气,研究成果主要体现在O3污染和前体物及气象要素的相互关系(安俊琳等, 2010; Zheng et al., 2010; Ran et al., 2012)。依然十分缺乏臭氧和其他氧化剂的关系及氧化剂对二次颗粒物生成速率的影响研究,且对大气氧化性垂直结构及区域差异影响等方面的研究仍十分匮乏。
目前,很多研究以OH 浓度来衡量大气氧化性,并在自由基测定技术方面取得一些重要成果(任信荣等, 2001; 邵敏等, 2004),总氧化剂Ox(近似等于O3+NO2)也常用于反映大气氧化能力的变化。程艳丽等(2008)使用还原性污染物在大气中的准一级化学反应速率常数定量表征大气氧化性,并尝试用于珠三角大气氧化能力数值模拟研究。Hofzumahaus et al.(2009)的研究发现,我国珠三角大气OH 存在与传统光化学理论不同的未知再生机制,可极大地提升痕量气体的降解速率,但并未伴随臭氧光化学生成率的增加,而且对细颗粒物的影响尚不明确。大量文献报道,数值模式对我国现阶段重霾污染过程颗粒物浓度的模拟结果普遍低于实测值,对大气氧化能力考虑不足或机制缺失,可能是造成不同程度低估颗粒物中二次成分的生成速率和产率的重要原因。大量的NOx排放极大地促进了SO2(S-IV)向硫酸盐(S-VI)的转化,气态NO2(N-IV)及存在于液滴中的HNO3(N-V)和颗粒物中NO3-(N-V)等亦可充当S-Ⅳ的氧化剂,矿尘和水成为非均相反应的催化剂及反应界面(王跃思 等, 2014; He et al., 2014; Wang et al., 2016)。
Gao et al.(2016)将这一氧化过程机理SO2(S-IV)→SO42-(S-VI)参数化后加入到数值模式研究中,使硫酸盐的产率提高了15 倍,并与观测结果高度吻合;香港的研究也证实上述反应途径对夜间硫酸盐的贡献高63%(Chow et al., 2016)。由于我国改革开放初期经济的迅猛发展,燃煤和燃油污染超量排放,大量还原性和氧化型污染物同时混存于大气中,相互间发生了非常复杂的非线性化学反应。氧化反应与中和反应相互促进,物理吸湿增长与二次化学过程相互耦合反馈,一度表现为重污染频繁突发。我国大气氧化性、氧化能力演变和氧化过程、路径及其污染形成的方式均显著区别于其他国家或地区,需要系统量化研究。
长期以来,国外学者大多将研究重点集中在大气氧化剂对自然源VOCs 的清除作用,而我国尚未完全解决大气氧化对SO2、NOx和NH3等无机污染气体的氧化去除机制问题,对人为源VOCs 的氧化机理研究也刚刚起步,鲜见大气氧化能力和空气质量水平的相互关系定量研究,对大气氧化能力缺乏具体的定量表达方式,难以提出通过调控大气氧化能力控制大气复合污染的理论依据。为此,本文作者近年来在国家和地方多项科研项目的资助下,开展系列AOC 与空气质量的定量关系研究,并在AOC 量化研究方面取得了突破性进展。主要表现在:建立了AOC 研究方法体系,阐明了AOC 的内涵,构建一套定量表征指标体系;完善了大气氧化剂收支平衡理论,并将其模块化引入了数值模式,提高了数值模式对AOC 的模拟能力;建立了大气氧化剂水平输送、垂直交换和局地生成过程的诊断方法,揭示出残留层大气存储的氧化剂或污染物,可直接对近地面环境空气质量造成不利影响。结果表明,AOC 与二次污染存在明确的量化关系,基于AOC 调控大气O3和PM2.5复合污染具有可行性。本文则重点介绍构大气氧化能力表观指数(AOIe,evaluation index of AOC)和 潜 势 指 数(AOIp,potential index of AOC)的建立方法和理论基础,并通过二者归一化指数闭合研究,探讨“丢失”的大气氧化过程或通道;大气氧化指数随污染程度不同的变化及其时空分布;AOC 主控者大气OH 自由基与其典型储库分子HONO 的收支关系;阐释NO2的光解系数[J(NO2)]与AOIp 的内在关系,使用AOIp 预测我国大气臭氧污染潜势格局;与其他化学反应量化指数对比,探讨其准确性、普适性和实用性。
2 大气氧化能力的内涵和量化指数的建立
本文作者从微观动力学和宏观热力学两个方面探讨了AOC 的内涵,建立了定量表达式,并开展了闭合示范研究,取得了突破性进展(Liu Z R et al., 2021; Yang et al., 2021)。
2.1 大气氧化表观指数(AOIe)的建立
化学反应的本质是原子或原子团重新拆分组合,形成新的物质分子;而大气中的氧化反应全称应是“氧化—还原”反应,其内涵是必须存在元素化合价的变化,本质是原子间有电子得失。失电子者被氧化,称为还原剂,大气中最常见的还原剂为还原性污染物SO2、NO、CO 和异戊二烯等烯烃类有机物;得电子者被还原,称为氧化剂,大气中最常见的氧化剂污染物为O3,氧气为大气中量最大的氧化剂但不是污染物,自由基OH、HO2和NO3等是活性最强的氧化剂,虽然浓度低,但活性极高,作用极大。在认知大气氧化性内涵基础上,作者从化学反应热力学原理出发,推算出一次排放的还原性前体物向氧化性二次污染物转化过程中得失电子的总摩尔数量,用以定量表征大气氧化能力,从宏观角度构建了一个大气氧化能力量化表达式,定义为“表观大气氧化指数”,缩写为“AOIe”。“AOIe”这个指数的含义是指这个大气氧化指数(AOI)可通过实验(experiment)获得,具有一定的经验性(empirical)。其计算相对简单,只关心一次排放的污染物氧化降解变成二次污染物过程中得失的电子摩尔数量,通过污染物中心元素化合价的前后变化即可计算获得,而不关心反应速度、转化途径和通道。其表达式为
其中,AOIe 代表参与大气氧化—还原反应(包括均相和非均相)所有化学物种得失电子摩尔量总和,单位可选用“μmol/m3”;fe1(S-IV→S-VI)代表大气中四价硫化合物(主要包括二氧化硫、亚硫酸或亚硫酸盐)转化为六价硫化合物(主要是硫酸或硫酸盐)过程中得失电子的总摩尔量(或当量,下同);fe2(N-II,III,VI→N-V)代表二价氮(NO)、三价氮(HONO或亚硝酸盐)、和四价氮(NO2)转化为五价氮(主要是N2O5、HNO3和硝酸盐)或是之间相互转化得失电子的摩尔量;fe3(O2→O3)代表大气中的氧气(O2)通过任何途径转化为臭氧(O3)过程中得失电子的总摩尔量;fe4(VOC→OVOC,SOA)代表VOCs在大气中被氧化生成含氧挥发性有机物(OVOCs)或氧化性二次有机气溶胶(SOA)得失电子的摩尔数当量,有机物中增加1 摩尔氧(O)计算为交换2 摩尔电子,如,甲烷(CH4)氧化成为甲醛(HCHO)计为2 摩尔电子转移; ε表示大气中其他痕量物种参与大气化学反应得失电子的摩尔量,如重金属、半挥发或难挥发有机物等。
对SOA 生成过程得失电子的表征,假定一次排放的挥发性有机物均为不含氧化合物,而氧化产物SOA 中含氧量的多少即代表其氧化程度,因此SOA 生成过程的得失电子量可由下式计算:
其中,氧碳质量比(O/C)由气溶胶飞行时间质谱仪直接监测获得,而SOA 主组成质量浓度可由HR-TOF-MS(高分辨率飞行时间气溶胶质谱)监测得到的有机物质谱信息结合正交矩阵因子模型(PMF)解析获取。
AOIe 这一指数的特点是:只关注结果而忽略过程。通过实测的污染物浓度拆分出氧化—还原反应产物质量浓度,计算出反应过程得失电子摩尔数量。AOIe 易获得,准确度高,能够反应客观事实;容易与数值模式对接,也可用于预测;但计算时,在一/二次污染物的分类上方面存在经验判断,尤其是对有机物的氧化产物判断要依赖气溶胶飞行时间质谱这类高端仪器设备的分类探测数据。
2.2 大气氧化潜势指数(AOIp)的建立
受到国内外学者早期研究工作(程艳丽等,2008; 林云萍和赵春生, 2009)的启发,作者从化学反应动力学基本原理出发,定量一次污染物在大气中准一级化学反应氧化去除速率加和来表征大气氧化性,建立了另一个AOC 评估指标,定义为大气氧化潜势指数(AOIp),其表达式为
其中,AOIp 代表各还原性污染物种在大气中通过准一级氧化反应去除速率总和,单位可选用“ppb/h”(1 ppb=10-9)或者“moleculer cm-3s-1”分子数;[Cj]表示第j种还原性污染物(SO2、NO、CO、CH4和NMHCs 等)的浓度;[Xi]表示第i种氧化剂(OH、HO2、NO3和O3等)的浓度;kij表示第j种污染物与第i种氧化剂反应的准一级化学反应速率常数,目前,大多数学者选择采用MCM(Master Chemical Mechanism)光化学机制中的速率常数;ξ表示未测定的活性还原性污染物消耗氧化剂的去除速率,可通过计算的OH 反应活性(KOH)与实测的OH 总反应活性的差值来表征。由于观测技术的发展,与国内外早期研究不同,目前AOIp 计算所需大多数物种和浓度,均可通过外场观测实验获取(Yang et al., 2021)。尤其是OH、HO2和NO3自由基浓度观测技术,中国学者目前均取得了突破性进展(Lu et al., 2019; Yan et al.,2021)。
AOIp 这一指数的特点是:计算式中的“k”值大多数都可从文献中获取,还原性反应物主要物种可通过观测获取。自由基氧化剂观测数据目前并不丰富,但可通过相应经验模型计算获得。因此,AOIp 计算难度不大,更容易与数值模式对接,容易扩展应用。但总的来说,AOIp 计算的只是一种潜势,一种可能性,并不一定真实发生,这一点与AOIe 截然不同,这也是二者可以进行闭合研究,寻找“丢失”大气氧化过程的理论基础。
2.3 大气氧化性指数AOIe 和AOIp 的闭合研究方法
本文作者从大气氧化反应的结果(AOIe)和大气氧化的潜势(AOIp)两个方向去描述同一个大气特性——AOC,试图从分析二者的差异寻找未被发现或是不明确的大气氧化反应通道或是新机制。但两个指数的量纲不同,为了方便比对研究,作者利用各自氧化进程的百分比法进行了无量纲归一化处理。以研究AOC 日变化为例,具体做法为:计算出当天AOIe 或AOIp 的小时均值(也可以是分钟或秒均值),然后计算出每个小时均值与当日最大值(或中值或平均值)的比值作为归一化的数值,简写为“N_AOIe”和“N_AOIp”。如果评估或是预测一个污染过程AOC 的变化形式,可参照上述处理方法,先找出过程最大值(或中值)进行归一化处理。
2.4 大气氧化能力指数的实际测算
为了研究这一大气化学理论问题,借助国家重点研发计划项目“区域大气氧化能力与空气质量的定量关系及调控原理”,于2017~2020 年,在中国科学院大气物理研究所铁塔分部实验站和河北香河实验站开展了为期4 年的观测实验。
2.4.1 北京铁塔观测站和河北香河观测站AOIe 统计日变化
根据公式(1)和公式(2)以及北京铁塔观测站和河北香河观测站的外场观测数据(表1),对观测期间的AOIe 进行了计算。本文所使用的夏季观测时间段为2018 年6 月1 日至7 月15 日,冬季观时间段为2018 年12 月1 日至2019 年1 月15 日。计算用到的观测数据包括臭氧、一氧化氮、二氧化氮,颗粒物化学组分[硫酸盐、硝酸盐和二次有机气溶胶(SOA)]以及颗粒物氧化状态(O∶C)。SOA 来自气溶胶飞行时间质谱数据结合正交矩阵因子模型解析得到,O∶C 来自气溶胶飞行时间质谱数据。

表1 计算夏季和冬季AOIe 所用的香河观测站和北京铁塔观测站的观测数据Table 1 The observations used for calculating the AOIe[evaluation index of AOC (Atmospheric Oxidation Capacity)] values in summer and winter obtained from the Xianghe station and the meteorological observation tower station in Beijing
北京城区AOC 由两部分组成,通过气相均相氧化—还原反应生成二次污染物计算出的大气氧化指数AOIe_G 统计日变化(观测期所有日变化过程的平均)形式为午间单峰型(图1b、e),准确表达出均相大气光化学过程每日中午前后最为剧烈;通过颗粒态非均相氧化—还原反应生成二次污染物计算出的大气氧化指数AOIe_P 统计日变化形式则为双峰型(图1a、d),但夜间的峰值远高于白天,准确表达出非均相大气化学过程往往发生在夜间,而白天的气—粒转化非均相化学过程较弱。从AOC 日变化总体形式TAOIe(图1c、f)分析,北京城市大气AOIe 总体日变化形式由均相大气光化学主控,但非均相化学过程不可忽视,尤其是秋冬季。较高的辐射强度和气温是驱动白天气相均相光化学过程的主动力,而夜间较高的相对湿度造成的较高的气溶胶含水量加之较低的混合层高度,造成了夜间较为强烈的非均相化学反应。图1a、d 午间出现的次峰值,准确反应出白天强烈的气相化学反应过程均会伴随着气—粒转化发生,往往是新粒子的生成过程。
河北香河秋冬季和春夏季大气表观氧化指数日变化(图2)总体变化形式与北京城区相似,尤其是春夏季几乎相差无几,但秋冬季表现出颗粒态非均相过程对TAOIe 的贡献比例更大。但无论是香河郊区还是北京城区,与秋冬季节不同,春夏季AOIe_P 峰值出现在上午08~10 时(北京时,下同),并不完全与交通早高峰时间段吻合。分析发现,这一时段,颗粒物中硝酸盐占比往往上升最快,其中硝酸铵比重最大,并与早间大气中气态浓度峰值相一致。关于北京城市氨气早高峰原因的研究结果为,汽车尾气排放的氨气被水汽吸收,夜间富集于植物叶片表面的露水中,随着早间气温升高,露水蒸发释放气态氨,形成氨气早间高值(Gu et al.,2022)。碱性氨气的早间高值无疑增加了对气态硝酸的中和,引发了颗粒态硝酸铵(NH4NO3)的生成。酸碱中和反应并没有元素化合价的变化或是电子转移,对AOIe 没有贡献,但却促发了四价氮N(IV)-NO2向五价氮N(V)-HNO3氧化—还原反应平衡的向右移动。高氧化态产物增加,电子转移数目增多,AOIe 值升高。研究揭示了华北大气中过量的氨释放,中和了氧化过程产生的酸性物质,一方面增加了二次颗粒物的产率,另一方面引发的大气氧化性增强,也可能增加了臭氧的产率,造成了以PM2.5和臭氧为代表的大气复合污染增加。因此,通过降低氨排放降低大气复合污染,表观上是控制了大气中和反应造成的颗粒态污染增加,实际上对AOC 具有间接的降低作用,也会同步降低气态臭氧污染的上升。
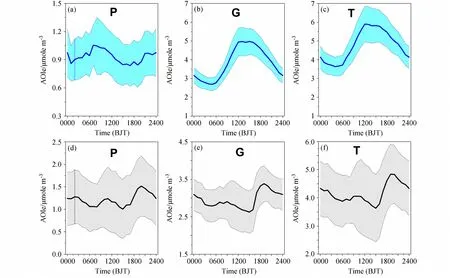
图2 同图1,但为河北香河观测站AOIe 日变化。Fig.2 As in Fig.1, but for diurnal variations of AOIe at the Xianghe station in Hebei Province.
本研究表明,大气中的Ox(O3+NO2的摩尔浓度)是北京城区AOIe 的最大贡献者,夏季对TAOIe 最高值的贡献为93%,冬季为84%。换句话说,颗粒态非均相过程对TAOIe 的贡献,夏季为7%,而冬季高达16%。在以往的大气氧化能力研究中,非均相大气化学过程往往被忽视,从而在数值模式模拟二次颗粒物产率时,往往会低估,造成对颗粒物污染浓度的预估严重偏低,同时也可能造成对臭氧浓度模拟的过大偏差。
2.4.2 北京铁塔观测站和河北香河观测站AOIp 统计日变化
根据公式(3)以及北京铁塔观测站和河北香河观测站的外场观测数据(表2),对观测期间的AOIp 进行了计算。本文所使用的夏季观测期时间段为2018 年6 月1 日至7 月15 日,冬季观测期时间段为2018 年12 月1 日至2019 年1 月15 日,其中计算用到氧化剂包括臭氧、OH 自由基和NO3自由基,其中OH 自由基和NO3自由基由经验公式计算得到;还原性污染物观测数据包括一氧化碳、一氧化氮、二氧化氮、二氧化硫以及68 种挥发性有机物,详细的物种信息及其与主要氧化剂的反应速率常数见Liu Z R et al.(2021)的表S1。表2 显示了北京铁塔观测站和香河观测站夏季和冬季观测期间主要氧化剂和还原性污染物的统计结果。

表2 同表1,但为计算AOIp 所用的观测数据Table 2 As in Table 1, but for the observations used for calculating the AOIp [potential index of AOC] values
北京和香河夏季和冬季的大气氧化能力潜势日变化均呈现午间单峰型[Liu Z R et al.(2021)的图5],无论是夏季还是冬季,其白天总量变化均由分量AOIp_OH 主控,而夜间则由AOIp_O3主控,但NO3的氧化作用也不可忽视,尤其是夜间城市上空(Yan et al., 2021)。北京城区春夏季观测期OH、O3和NO3自由基表征的大气氧化能力分别为2.38×107molecules cm-3s-1、3.10×106molecules cm-3s-1和4.89×105molecules cm-3s-1,分别占总AOIp 的76.7%、22.2%和1.0%。城区秋冬季观测期OH、O3和NO3自由基表征的大气氧化能力分别为0.50×107molecules cm-3s-1、0.22×106molecules cm-3s-1和0.02×105molecules cm-3s-1,分别占总AOIp 的95.9%、4.1%和<0.01%。北京郊区春夏季观测期OH、O3和NO3自由基表征的大气氧化能力分别为2.50×107molecules cm-3s-1、3.74×106molecules cm-3s-1和4.55×105molecules cm-3s-1,分别占总AOIp 的85.6%、12.8%和1.6%。郊区秋冬季观测期OH、O3和NO3自由基表征的大气氧化能力分别为0.81×107molecules cm-3s-1、0.19×106molecules cm-3s-1和0.05×105molecules cm-3s-1,分别占总AOIp 的80.9%、18.9%和0.2%。无论是城区还是郊区,白天AOIp 的主要贡献者均为OH,其次为O3,其氧化的主要燃料是烯烃(>95%),而芳香烃贡献了其余部分;夜间AOIp的主要贡献者均为O3,由于OVOCs 浓度较低,NO3自由基贡献较小。相比较夏季,城区OH 自由基对AOC 的贡献在冬季进一步上升,而城郊则正好相反,冬季相比夏季O3表征的大气氧化能力有所上升,这与氧化剂的主要燃料—挥发性有机物的物种组成在城区和郊区存在较大差异有关。
AOIp 还原剂分量贡献最大者为烯烃,其次为芳香烃、OVOCs 和烷烃,氟氯烃贡献最小[见Liu Z R et al.(2021)的图6]。综合分析来看,对大气氧化能力潜势影响最大的是大气中的OH 自由基与烯烃、芳烃和烷烃的一级光化学反应,需要指出的是此处计算OVOCs 的贡献时并未包含甲醛和乙醛等的观测结果,OVOCs 对AOIp 的贡献可能存在低估,这从2.4.3 节AOIe 与AOIp 的闭合分析可以看出上述OVOCs 物种对日间AOIp 贡献较大;同时本研究发现,OVOCs 近年来对大气氧化能力的作用越来越强,已经达到了十分显著的程度,这与最近在珠三角(Wu et al., 2020; Wang et al., 2022)等地报道的结果一致。
必须指出的是,本文定义的AOIe 和AOIp 算法公式,均是一个开放的体系,可根据观测站点或区域的大气污染实际状况增减大气二次污染物、氧化剂或还原剂的种类,但必须以客观实验数据为依据,对于无法获取的化学量,如RO2自由基,暂不纳入计算体系,但不影响二者的归一化对比研究工作的开展和研究结果的相对客观性。
2.4.3 AOIe 与AOIp 的闭合研究及其科学价值
根据定义,AOIe 只关注最终结果,忽略中间过程,优势为全面考虑了均相与非均相氧化反应过程,因此,更加接近客观的AOC 量化表达。这是本文作者独创的对AOC 一种全新量化表达方式。AOIp 仅考虑气相均相过程,而且是大气氧化反应发生潜势的一种表达方式,并不能反映出大气氧化反应的最终结果,其优势是可以对大气氧化性进行预测。理论上,二者应能趋于闭合。为了分析导致AOIe 和AOIp 日变化差异的原因,研究者挑选了夏季和冬季外场观测案例,评估可能忽略的反应途径和活性VOCs 物种对AOIp 的影响。
正如前文所述,由于AOIe 和AOIp 表征大气氧化性变化过程依据的化学反应途径和时间尺度各有不同,因此,二者的量纲不同。为了对二者闭合分析以寻求可能“丢失”的大气氧化反应或是途径,本文作者通过指数的每一个小时均值除以日最大小时值将上述两个AOC 指数归一化,转换为无量纲的百分比变化单位,简写为NAOIe 和NAOIp。从图3a、b 可以看出,城区与郊区夏季NAOIe 和NAOIp 各自的日变化差别不大,冬季有一定差异,但不十分显著,但两指数之间相比,除了中午,其他时间段呈现较大差异,尤其是夜间,城区NAOIe和NAOIp 差异约为83%~96%(图3a),而城区为80%~97%(图3b);冬季,两者的差异也同样出现在夜间,城区相差98%~99%,城区相差94%~96%。总体而言,两站点、两季节NAOIp出现峰值的时间均在午间,表现出气相均相光化学过程对AOC 潜势的主控;两站点、两季节NAOIe出现峰值的时间发生了显著变化,夏季其与NAOIp相同出现在中午,而冬季则出现在夜间,表明这两个AOC 指数在表征大气氧化能力方面尚不能完全闭合,显示出我们对大气氧化过程和机理的认知存在盲区。显然,冬季AOIe 的最大贡献者来自非均相大气氧化过程,而这一点在以往的大气氧化能力研究中往往被忽视,因此,有必要深入探讨。
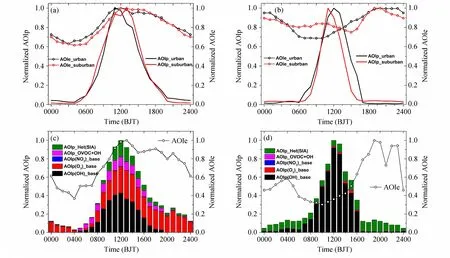
图3 北京铁塔观测站和河北香河观测站(a)夏季观测期(2018 年6 月1 日至7 月15 日)、(b)冬季观测期(2018 年12 月1 日至2019年1 月15 日)归一化的AOIp、AOIe 日变化,(c)夏季、(d)冬季典型污染个例中某一天修订后的NAOIp(归一化的AOIp)分量组成(柱状)、NAOIe(归一化的AOIe)分量组成(带圆圈的线)的日变化叠加。图c、d 中的某一天分别为2018 年7 月4 日臭氧污染日、2019 年1 月12 日颗粒物污染日。Fig.3 Diurnal variations of normalized AOIe (NAOIe) and normalized AOIp (NAOIp) in (a) summer observation period (from 1 June to 15 July 2018) and (b) winter observation period (from 1 December 2018 to 15 January 2019), cumulative plots of daily variations for NAOIp components(bars), NAOIe components (lines with circles) in typical pollution cases during (c) summer (typical ozone pollution day on 4 July 2018) and (d) winter(typical particle pollution day on 12 January 2019) at the meteorological observation tower station in Beijing and at the Xianghe station in Hebei Province.
在理解AOC 内涵的基础上,计算AOIp 时考虑了主要氧化剂与OVOCs 的一级动力学均相氧化反应与多路径硫酸盐和硝酸盐的非均相化学反应。具体做法如下:首先,使用了基于MCM3.3.1 版本的箱式模型(F0AM)来模拟OVOCs 的贡献。选择夏季臭氧污染天(2018 年7 月4 日)和冬季颗粒物污染天(2019 年1 月12 日)作为案例的模型结果,表明甲醛、乙醛和苯甲醛是贡献OH 消耗的重要物种,三者贡献的OH 消耗率可达10 ppb/h,占OH 总消耗率的近30%,但三者对冬季OH 消耗量贡献相对较低,OH 消耗率约为2 ppb/h,约占OH 总消耗率的12%。因此,将OVOCs 种类补充进AOIp 的计算,可减少对AOC 的低估。其次,为了评估非均相过程对AOC 的贡献,研究者使用多相化学箱模型(RACM-CAPRAM),并耦合了四种非均相硫酸盐生成机制(气溶胶相中H2O2、O3、NO2和TMI 催化氧化途径)来模拟硫酸盐和硝酸盐形成的非均相机制。为了与OVOCs 模拟便于比较,同样选择了夏季和冬季同时间案例。多相化学箱模型模拟结果显示,在冬季案例中,气相均相反应的贡献仅占硫酸盐生成的45%左右,而非均相反应的贡献超过50%,并主导了硫酸盐增长。Ye et al.(2021)的研究也指出华北冬季重霾期间非均相反应途径对硫酸盐二次生成的贡献可达70%左右,证实了如果不考虑非均相机制,AOIp会大幅低估AOC。对于硝酸盐,气相均相反应贡献了95%,在污染高峰阶段N2O5水解途径贡献了5%。以往的研究指出由于污染高峰阶段高浓度NO的存在,使得近地面的N2O5浓度接近于零,因而N2O5水解途径对硝酸盐生成的贡献可以忽略不计(Wang et al., 2018),与本研究模拟的结果一致。
图3c、d 显示了改进的AOIp 的日变化特征,计算中加入了二次无机气溶胶(SIA)形成的液相和非均相生成过程。在计算中添加OVOCs 活性物种和非均相氧化过程在很大程度上改善了AOIp 对AOC 的低估,从另一个角度反映出以往AOC 研究对含氧有机物的贡献考虑不足,而对多介质的非均相过程更是缺乏考虑,尤其是颗粒物污染严重的冬季。
图3c 显示,夏季重污染过程经过补充均相OVOCs 和非均相SIA 氧化过程计算出的NAOIp与相应的NAOIe 更加接近,尤其是在09~13 时二者几乎重合,但下午直到夜间,差异仍然显著,有可能是因为SOA 对AOC 的贡献计算的欠缺。图3d显示,冬季重污染过程经过补充均相OVOCs 和非均相SIA 氧化过程计算出的NAOIp 发生了显著性变化,夜间峰值与相应的NAOIe 接近重合,但白天午间峰值出现较大偏差。原因可能是冬季重污染过程光线弱、温度低,不利于光化学反应的发生,大气中的氧化—还原反应几乎以非均相的二次颗粒物生成过程主导。换句话说,AOIp 计算的仅是一种均相大气光化学反应潜势,如果光热条件具备不充分,这种反应真实发生的可能性就会大幅度降低,此时,大气氧化性主要由二次颗粒物生成的化学过程体现。这一点,也是以往相关研究中被忽略的一个大气氧化过程或途径。需要强调的是,AOIp 和AOIe 的计算是一个开放的体系,开展归一化闭合研究的目的是为了寻找或是确认大气中“丢失”的大气氧化过程或是通道,“填补”二者之间差异是本研究的目标和动力。目前,AOIp 和AOIe 产生差异的原因之一可能是基于参数化方法计算的自由基浓度有误差,另一方面原因是AOIp 计算中仍未全面考虑SOA 的非均相生成;二次污染物的区域传输或是排放源高氧化态污染直排均可造成局地AOIe 计算值虚假偏高。进一步研究不仅需要大气自由基的直接外场观测数据和厘清一次/二次各类污染物来源,还需要进一步确认模式计算中采用各种反应动力学常数的准确性,考虑多界面、多层次和多级大气氧化反应。
3 大气氧化能力研究的拓展与应用
3.1 不同颗粒物污染程度下AOI 的演变
将北京城市和香河郊区站观测时段污染状况按照PM2.5日均浓度的环境空气质量标准分成优、良、轻度、中度及以上四种状况,可得到AOI 随颗粒物污染加重变化趋势。随着污染程度的加重,无论是夏季或是冬季、城区或是郊区,总体上AOIe 均随之升高,但AOIp 的变化有所不同[见Liu Z R et al.(2021)的图9]。夏季随着颗粒物污染的增强,AOIp 出现先升高后下降的趋势,表现出较高的颗粒物浓度对气相均相光化学过程产生了抑制作用。京津冀区域,PM2.5阈值区间为75~115 μg/m3。以往研究认为,我国北方大气中PM2.5浓度达到80 μg/m3就会显著影响大气水平能见度(Zhao D D et al.,2021),也就是说中度及以上污染太阳辐射显著降低,大气均相光化学反应减弱,但不影响非均相大气化学过程的继续加强。冬季随着颗粒物污染的加重,AOIp 整体下降的趋势,几乎与AOIe 的变化完全相反,说明我国北方区域冬季大气氧化能力主要受非均相化学过程所控制。
3.2 大气氧化能力与大气中的气态亚硝酸收支
大气中的气态亚硝酸(HONO)中心化学元素为N 元素,价态为中间价态+3 价,所以HONO 既可以作为大气中的氧化剂,也可作为还原剂。HONO是对流层大气HOx(HOx=OH+HO2)的重要来源,是影响大气AOC 变化的重要要素之一。有研究认为,白天HONO 光解对OH 自由基的贡献可高达60%以上,在加速气相化学反应的同时,也可能导致硝酸盐和SOA 的爆发性增长。但当前针对HONO 的来源尚未完全厘清,尤其是其非均相形成过程,探索其来源已成为大气化学领域的难点和热点。针对上述问题,本文作者基于AOIe 的闭合研究思路,以HONO 实验观测的与盒子模型模拟的AOIp 的差异性作为收支平衡研究的突破口,两条路径交叉耦合,确立了适用于北京城市复杂大气环境下的HONO 生成、降解机制的闭合研究方法,量化了HONO 生成对大气关键氧化剂HOx的贡献。
研究结果显示,北京城区白天HONO 主要来源于气相均相反应、颗粒物表面光增强非均相反应、机动车尾气直排和地表面光增强非均相反应生成;夜间则来自气相均相反应、大气NO2在地表面或颗粒物表面的非均相反应以及机动车直排[见Liu J Y et al.(2021)的图5]。夏季白天HONO 主要来自颗粒物和地表光增强非均相反应、气相均相反应、机动车直排和硝酸盐颗粒的光解,其平均贡献分别为34.4%、22.0%、18.5%和8.4%;夜间,HONO主要来源于NO2地表和颗粒物表面的非均相化学反应、气相均相反应及机动车直排,其贡献分别为42.0%、30.1%和12.0%。冬季北京大气HONO来源夜间和白天没有显著区别,颗粒物表面和地表光增强反应及机动车直排是三种主要来源,其日间的平均贡献分别为29.7%、24.9%和20.4%,而夜间的平均贡献分别为25.0%、17.5%和27.9%。
以往研究HONO 的来源时往往仅考虑气相均相反应,大气中HONO 浓度的低估造成关键氧化剂OH 自由基浓度的显著低估,从而造成AOC 的严重低估。耦合HONO 新增源后,尤其是非均相化学源,使OH 自由基的峰值平均浓度在夏季提升了39.7%,而冬季则提升了212.5%。与OH 自由基浓度变化相比,HO2和RO2自由基浓度变化较小。耦合HONO 新增源后,HO2自由基的峰值浓度分别提升了20.0%(夏季)和195.7%(冬季)。RO2自由基的峰值浓度分别提升了8.3%(夏季)和240.7%(冬季)。HONO 新增源的加入,使冬季AOI 值提升更显著,表明MCM 机制对冬季AOC低估较为严重,迫切需要在数值模型中充分考虑HONO 的非均相来源(Liu J Y et al., 2021)。
3.3 AOIp 用于全国大气臭氧污染潜势预测
本文作者利用在华北区域不同背景条件(城市、乡村)的光化辐射通量、地基太阳辐射分波段要素的同步观测数据,结合辐射传输模式建立了适合于华北区域光化辐射通量重构的参数方案,并通过了不同气候带的适用性检验,结果显示该参数化方案能够较好地应用于我国。利用中国生态研究网络(CERN)联网观测的长时间紫外辐射数据,结合参数化方案利用辐射传输模式实现了全国光化辐射通量的重构,最终形成了全国光化辐射通量数据集。利用光化辐射通量和观测的污染气体建立了臭氧光化学生成表观潜势(AOIp_O3)测算技术。
研究表明,近地层大气中的臭氧主要来自于环境大气中NO2的光解产生的氧原子O(3P)与大气中占比21%氧气的化学反应,化学反应过程如下:
其中,hv 表示光能,M 代表空气中的N2、O2或其他分子介质,可以吸收多余的能量来稳定反应生成的O3分子。近地层大气中的臭氧主要汇是被NO滴定,此时大气臭氧浓度平衡方程可简写为
式中,k1和k2分别表示化学反应过程(4)和(6)的一级反应速率。k1大小取决于NO2的光解系数J(NO2),k2也接近于常数,因此,根据本文对AOIp的定义,臭氧生成潜势主要取决于J(NO2)、NO2及NO。挥发性有机物对O3生成潜势的影响,则可通过其光化学反应生成的RO2自由基对NO 的氧化,最终也是通过NO2的环境大气浓度反映到臭氧生成潜势,因此可以将J(NO2)作为臭氧生成潜势指标。
由于我国J(NO2)观测点很少,而且开展的时间较晚,但CERN 全国尺度长时间的紫外辐射观测为反演J(NO2)提供了基础。光解速率可由辐射传输模型模式计算获得。光解速率与紫外线辐射比值存在较稳定相关关系,因此少数学者基于建立的经验模型(Palancar et al., 2005; Trebs et al., 2009)开展J(NO2)的数据重建,但是该比值依赖观测点的气候、地理位置,致使经验模型在观测站点之外的气候区域适应性较低。利用对流层紫外和可见光辐射传输模式(Troposphere ultraviolet and visible radiation model,TUV)来计算J(NO2)是国内外使用最广泛的方案。由于模式计算需要的云、气溶胶相态等相关参数不易获得,在使用时多采样晴空无云假设,从而带来较大的计算误差(Madronich,1987; Toon et al., 1989; Kylling et al., 2003)。
研究结果表明J(NO2)和紫外辐射一样主要受到云、气溶胶的影响,到达地面的理想状况下的J(NO2)、紫外辐射都能够通过辐射传输模式精确获得。紫外辐射和J(NO2)观测值与晴空条件下模拟值的比值定义为云修正因子(分别表示为CMFUV和CMFJ),通过紫外观测数据和模式可获得紫外辐射的云修正因子(CMFUV),分析发现紫外辐射云修正因子与J(NO2)云修正因子存在较好的指数关系(Zhao S M et al., 2021),从而可利用紫外辐射云修正因子来计算J(NO2)云修正因子(CMFJ),然后利用CMFJ订正模式模拟的J(NO2),从而获得更高精度的J(NO2)数据。把河北香河和北京观测的J(NO2)和紫外辐射数据随机分为两组,一组用于重构方法的建立,另一组用于重构效果的验证。在重构过程中假设云、气溶胶对辐射和光解速率的影响是相互独立的,紫外辐射和J(NO2)的云修正因子用如下公式表示:
其中,UVc和Jc分别代表用TUV 模型计算的无云条件下的紫外辐射和光解速率,UVobs和Jobs分别代表紫外辐射和光解速率的观测值。分别将香河和北京的CMFUV和CMFJ进行拟合即可得到基于紫外辐射观测的CMFJ。
检验结果表明建立的重构方法能够获得较高精度的计算值,J(NO2)瞬时观测值与计算瞬时线性拟合回归系数为0.96,平均相对误差为6.5%(图4)。进行重构方法适用性检验,把北京站点建立的参数化方案应用于香河,重构方法的精度较北京本地有较小的下降,这表明该区站点建立的参数化方案可以较好地应用于该气候区域。

图4 2018 年7 月至2019 年10 月观测期间(a)北京铁塔观测站、(b)河北香河观测站J(NO2)模拟值与观测值的比较。红色线是线性拟合结果。MBE、RMSE、R2 和N 分别表示平均偏差、均方根误差、相关系数平方和样本数量。Fig.4 Comparison of simulated and observed photolysis rate of NO2 [J(NO2)] at (a) the meteorological observation tower in Beijing, (b) the Xianghe station during the observation period from July 2018 to October 2019.The red line is the linear fitting result.MBE, RMSE, R2, and N represent mean bias error, root mean square error, square of the correlation coefficient, and the number of samples, respectively.
利用上面的方法对长期的光解速率J(NO2)进行重构,2005~2019 年期间,北京地区J(NO2)同样呈上升趋势,年均增幅为1.19%,且上升趋势在2014 之后尤为明显;整个时段内J(NO2)的平均值为2.70×10-3s-1,明显小于香河地区;年平均最高值为3.07×10-3s-1(2019 年),最低值为2.42×10-3s-1(2008 年)。北京地区的紫外辐射总体也呈上升趋势,但年均增幅仅为0.31%。紫外辐射年均值为0.39 MJ m-2d-1,同样小于香河地区;年平均最高值和最低值对应年份与J(NO2)不同,最高值在2016 年(0.41 MJ m-2d-1),最低值在2006年(0.36 MJ m-2d-1)。北京地区J(NO2)的增幅与香河地区一致,J(NO2)比紫外辐射的增幅更明显。AOD 在2005~2019 年总体呈下降趋势,年平均值为0.82,平 均 每 年 下 降2.56%[见Zhao S M et al.(2021)的图5]。
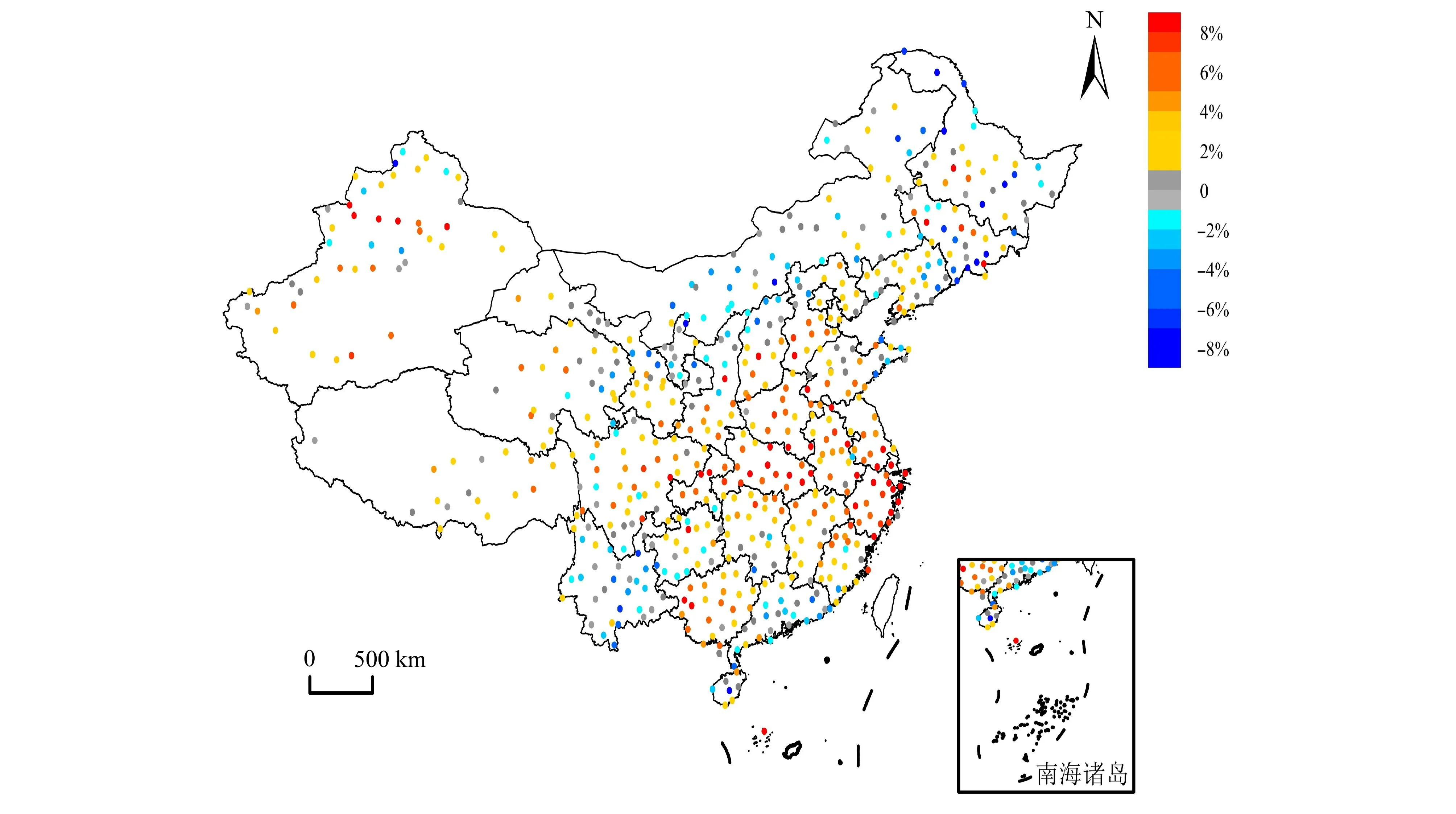
图5 2020 年与1961 年相比全国各站点J(NO2)变率。Fig.5 The J(NO2) change rate of all stations in China in 2020 compared with that in 1961.
利用重构获得1961~2020 年全国724 个气象站点紫外辐射数据(吴彤等, 2022),结合辐射传输模式获得全国CMFUV,通过CMFUV和CMFJ的拟合方程获得了全国范围不同区域的CMFJ;然后结合TUV 计算获得的晴空条件下的J(NO2),最后计算获得了J(NO2)数据。重构的60 年的J(NO2)的全国年均值为4.39×10-3s-1,其高值区主要分布在四川、贵州、重庆和湖南等地,而我国东北、华北、西北以及青藏高原地区的J(NO2)相对较小(图5)。2020 年全国绝大多数地区的J(NO2)与1961 年相比都呈现为增长的趋势,特别是中东部增长趋势更为明显。该数据为使用AOIp_O3测算我国各重点区域臭氧污染潜势提供了高精度的数据支撑。
3.4 大气氧化能力指数与其他氧化指数的对比
基于香河地区2019 年夏季O3及前体物挥发性有机化合物(VOCs)、NOx的连续观测,划分O3超标天与非超标天,依据经验公式并结合0-D 光化学箱模型(NCAR-MM)探究前体物对O3生成的影响,评价指标包括OH 反应性(KOH, OH reactivity)、臭 氧 生 成 潜 势(OFP, O3formation potential)和相对增量反应活性(RIR, relative incremental reactivity),同 时 与AOC 表 征 指 数(AOIp_G 和AOIe_G)比对研究,从气态氧化过程及气态氧化产物的角度,量化观测期间AOC 的变化。由图6a-c 可以清晰地看出,KOH、OFP 与RIR 类似,均表征出O3超标天的前体物VOCs 反应性高于非超标天,较高的前体物反应性对应着较高浓度的O3,其白天均值分别为10.4 s-1、331.7 μg m-3和0.91%/%。AVOCs(人为源VOCs)的活性占比均较高,超标天日均KOH-AVOCs、OFP-AVOCs、RIR-AVOCs 分别占65%(6.7 s-1)、86%(284 μg m-3)和72%(0.66%/%)。就VOCs 的不同类别而言,KOH中含氧VOCs(OVOCs)的份额(占KOH-VOCs的44%)超过异戊二烯(占KOH-VOCs 的35%),对O3的形成起着至关重要的作用,而烷烃、烯烃和芳烃所占比例均小于10%;OVOCs 与芳香烃有着较高的臭氧生成潜势,这两类VOCs 的OFP 占比均超过异戊二烯(14%),分别为48%、23%;而芳香烃在RIR 中占比最高(30%),异戊二烯、OVOCs 与烯烃的RIR 占比也较高,分别为28%、22%、16%。
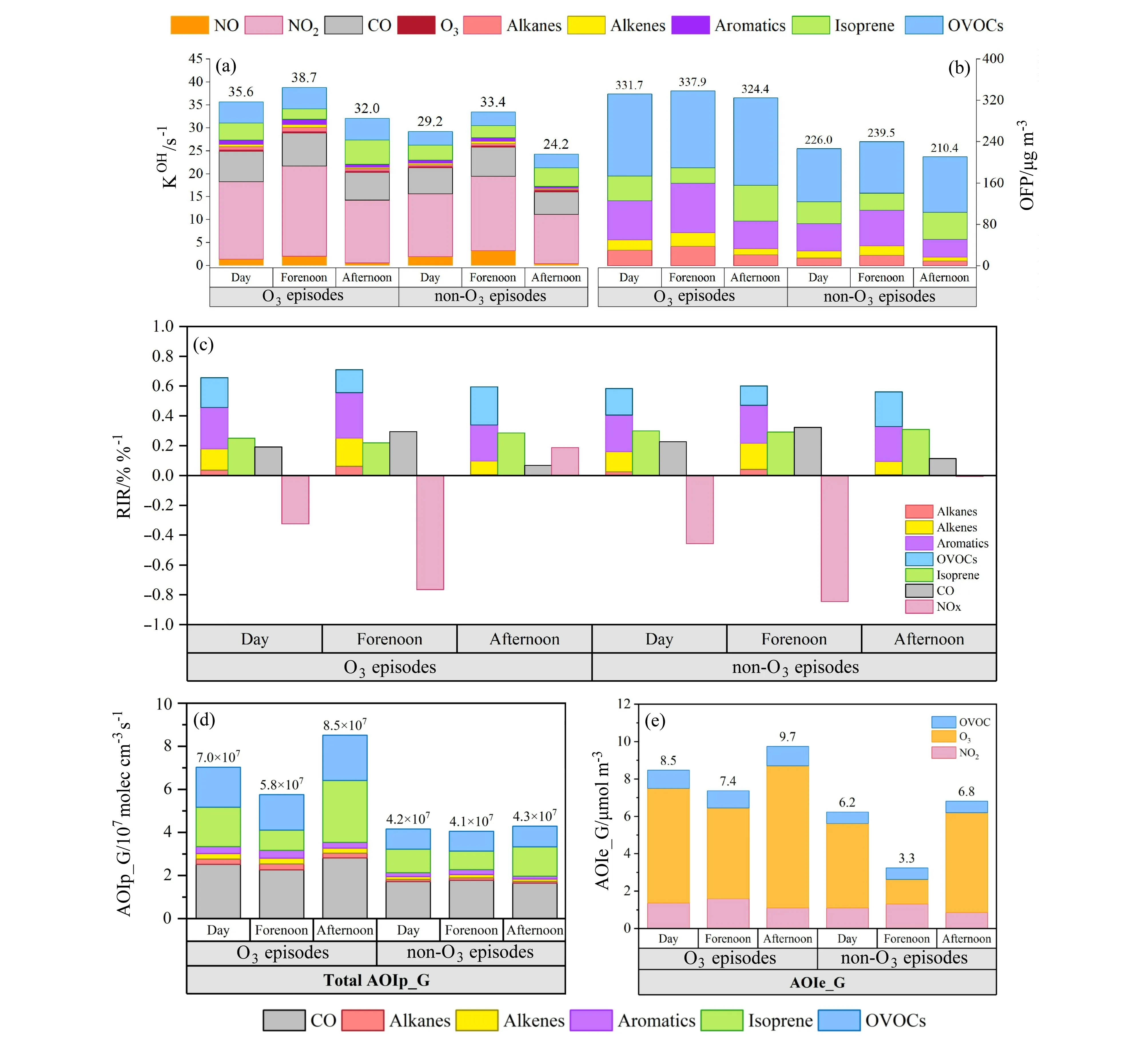
图6 2019 年夏季(6 月17 日至7 月1 日)香河地区白天(06~18 时)、上午(06~12 时)和下午(13~18 时)平均的(a)KOH、(b)OFP、(c)RIR、(d)AOIp_G 和(e)AOIe_G。Fig.6 (a) KOH (OH reactivity), (b) OFP (O3 formation potential), (c) RIR (relative incremental reactivity), (d) AOIp_G (potential AOC calculated by oxidation reaction rates), (e) AOIe_G (estimated AOC given redox electron transfer for oxidation) averaged in daytime (0600-1800 BJT), forenoon(0600-1200 BJT), and afternoon (1300-1800 BJT) in Xianghe area in summer (from 17 June to 1 July) 2019.
AOC 量化指数AOIp_G 和AOIe_G 变化形式具有一致性,O3超标天的AOIp_G 和AOIe_G 均显示出强于非超标天,日均值分别为6.7×107molec cm-3s-1和8.5 mol m-3,表征出较高的大气氧化能力可能促发O3污染的发生。总体上,上午时段臭氧前体物光化学活性表征指数KOH、OFP 和RIR均高于下午时段,AOIp_G 和AOIe_G 的变化与上述三个指数具有一致性,表明它们具有相同的内在驱动力。KOH、OFP、RIR 通常用于表征一次前体物通过大气中复杂的氧化还原反应对二次污染物O3的相对贡献,而大气氧化性指数AOIp_G 和AOIe_G 作为大气氧化能力的量化指标则更为全面,还囊括了最终生成其他二次污染物如SOA 的部分大气氧化能力。AOIe_G 从产物的角度出发,包含多种相态的反应过程,更接近实际的大气氧化能力,而AOIp_G 仅量化气态前体物被氧化剂氧化的潜势(可能性),仅考虑了气相反应部分所对应的大气氧化能力。
图7 显示,随着AOIp_G、AOIe_G 的增加,臭氧产生率P(O3)与O3浓度呈现上升趋势,表征出AOC 的增加伴随着O3光化学生成的加剧以及环境浓度水平的升高。RIR-VOCs 与RIR-NOx则呈现不同的变化趋势,表现出香河地区O3光化学生成整体处于VOCs 控制区。但同一天O3光化学敏感性会随着反应的进行而发生变化,前体物处于持续消耗的状态,得益于生物源VOCs 与气温正相关的排放率对VOCs 总浓度的补给。环境大气上午早间时段从富NOx条件向下午时段贫NOx的状态转变,O3生成敏感性也由VOCs 控制区转向VOCs与NOx的协同控制区。AOIp_G 和AOIe_G 可以较好地指示环境O3的污染水平,其中AOIe_G 与O3小时浓度呈现良好的线性相关。AOIp_G 增加表征出ROx氧化NO 的气相均相反应增加,臭氧产量增加,在贫NOx条件下,ROx之间的反应增强,部分AOIp_G 表征二次有机气溶胶产率的增加。AOIe_G 涵盖了多相反应过程,较好地捕捉了大气光化学过程,即二次产物的水平和变化;相比之下,AOIp_G 只评估了前体物气态氧化过程的可能性,并且以VOCs 氧化为主,因此表征出AOIp_G 和O3浓度之间的较为复杂的相关关系。

图7 2019 年夏季(6 月17 日至7 月1 日)香河地区白天(a)AOIp_G、(b)AOIe_G 与O3 小时浓度的相关性,(c)AOIp_G、(d)AOIe_G与O3 生成速率的相关性。Fig.7 Correlations between (a) AOIp_G, (b) AOIe_G and hourly O3 concentrations, between (c) AOIp_G, (d) AOIe_G and P(O3) (O3 production rate) in Xianghe area in daytime in summer (from 17 June to 1 July) 2019.
4 结论
在认知大气氧化性内涵的基础上,创新性地建立了AOC 的量化研究方法,构建出表观大气氧化能力指数AOIe 和大气氧化能力潜势指数AOIp,并通过二者归一化闭合量化研究,揭示出了非均相化学过程对AOC 具有显著性贡献,尤其在重霾污染或是在夜间高湿环境条件下,非均相大气氧化过程往往成为AOC 的主导。随着颗粒物污染的加重,表观大气氧化能力指数AOIe 随之呈线性增加,并且不受季节变化影响;大气氧化能力潜势指数AOIp 夏季有与AOIe 相似的变化趋势,但其冬季则出现了相反的情景,表现出AOIp 的变化受气象条件影响更大,主要是温度和辐射变化的限制。闭合思路用于大气最重要氧化剂OH 自由基储库分子HONO 研究,发现北京大气HONO 具有重要的非均相来源,由此阐释了广泛应用的MCM 机制对冬季AOC 低估的重要原因。大气氧化能力潜势指数用于预测我国大气臭氧污染潜势格局,发现AOIp_O3与J(NO2)直接相关,全国J(NO2)的年均值为4.39×10-3s-1,高值区主要分布在四川、贵州、重庆和湖南等地。与其他大气化学反应氧化性指标对比,本文构建的AOI 更具准确性、普适性和实用性,不但可评价已发生的污染过程AOC 的变化,亦可预测城市或区域重污染发生的可能性及其变化趋势。本研究在理论和技术上获得了双重突破,为基于AOC 调控原理的大气PM2.5和臭氧复合污染的协同控制策略制定提出了一条新的路径。
致谢 感谢项目研究过程中中国科学院大气物理研究所香河试验站、325 米铁塔维护工作人员和北京大学等诸多合作单位的鼎力支持。
猜你喜欢
农业灾害研究(2022年1期)2022-05-07
选煤技术(2022年1期)2022-04-19
能源工程(2021年2期)2021-07-21
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
环境保护与循环经济(2017年2期)2017-09-26
浙江农业学报(2017年1期)2017-05-17
杂文月刊(2017年24期)2017-03-03
化工进展(2015年3期)2015-11-11
浙江大学学报(工学版)(2015年1期)2015-03-01
