
单基因高血压的研究进展
2024-04-29 17:32张玉帕丽达阿布来提
心血管病学进展 2024年2期
张玉 帕丽达?阿布来提

【摘要】单基因高血压是一种重要的高血压遗传模式,它是一种由遵循孟德尔遗传的单一遗传变异引起的。目前,单基因高血压的临床发病率及发病机制尚不完全清楚,临床不易识别与诊断。现旨在对单基因高血压的分类、突变基因及机制特点、诊断治疗等方面进行系统阐述,以期为今后该疾病在临床的诊疗提供更多的参考。
【關键词】单基因高血压;孟德尔遗传;基因检测
【DOI】10.16806/j.cnki.issn.1004-3934.2024.02.000
Monogenic Hypertension
ZHANG Yu,Palida·Abulaiti
(Third departments of comprehensive internal medicine,The First Affiliated Hospital of Xinjiang Medical University,Urumqi 830000,Xinjiang,China)
【Abstract】Monogenic hypertension is an important genetic pattern of hypertension,which is caused by a single genetic variant that follows Mendelian inheritance. At present,the clinical incidence and pathogenesis of monogenic hypertension are not completely clear,and it is difficult to identify and diagnose clinically. The purpose of this paper is to systematically elaborate the classification,mutated genes,mechanism characteristics,diagnosis and treatment of monogenic hypertension,in order to provide more references for the clinical diagnosis and treatment of this disease in the future.
【Keywords】Monogenic hypertension;Mendelian inheritance;Genetic testing
高血压是全球心血管疾病罹患和过早死亡的主要原因之一,目前高血压的患病率仍呈上升趋势,而且由于其长期控制不佳,会造成多种靶器官损害,死亡率也日益增高[1]。单基因高血压(monogenic hypertension,MH)是由单一基因突变引起的高血压病,是高血压病的一种罕见类型。它的主要特征为早发严重高血压、酸碱代谢紊乱(最常见的是碱中毒和低钾血症)、低肾素水平以及阳性家族史[2]。其患病率不详,临床不易辨识与诊治,近年来,随着对MH发病机制的深入研究,对其突变基因的探究已成为新的热点,现简述MH的临床分类,重点综述MH在已知突变基因、诊断和治疗方面的进展。
1 MH的常见分类方法
MH的遗传形式源于盐皮质激素、糖皮质激素或交感神经通路中的功能获得或丧失突变。因此,根据突变基因的不同表型,MH有不同的临床分类方法。当突变引起盐皮质激素的功能改变时,我们常根据血清肾素和醛固酮水平对MH进行分类。首先,根据血清肾素水平可将其分为正常肾素MH和低肾素MH,而低肾素MH可根据醛固酮水平进一步分类:低醛固酮低肾素性高血压、正常醛固酮低肾素性高血压或高醛固酮低肾素性高血压。另一类是由于肾上腺素/交感神经功能突变引起的MH[3]。根据盐敏感性和醛固酮水平,MH合并低钾血症可分为三类:盐不敏感型高血压、盐敏感低醛固酮型高血压、盐敏感高醛固酮型高血压[2]。
同时,在临床上,MH也会用到其他的分类方法。肾脏和肾上腺是参与MH的两个主要器官,根据两个器官上的基因突变可分为:肾上腺突变型高血压和肾远曲小管、集合管突变型高血压[4]。另外,MH根据病理生理机制的不同可以分为五类:(1)醛固酮合成过度;(2)肾上腺类固醇代谢和作用失调;(3)远端小管中钠和氯转运体的过度活动;(4)肾上腺素能过剩;(5)血管平滑肌增生[5]。不论其如何分类,所有的MH都具有高血压这一共同临床表现。
2 MH的常见亚型
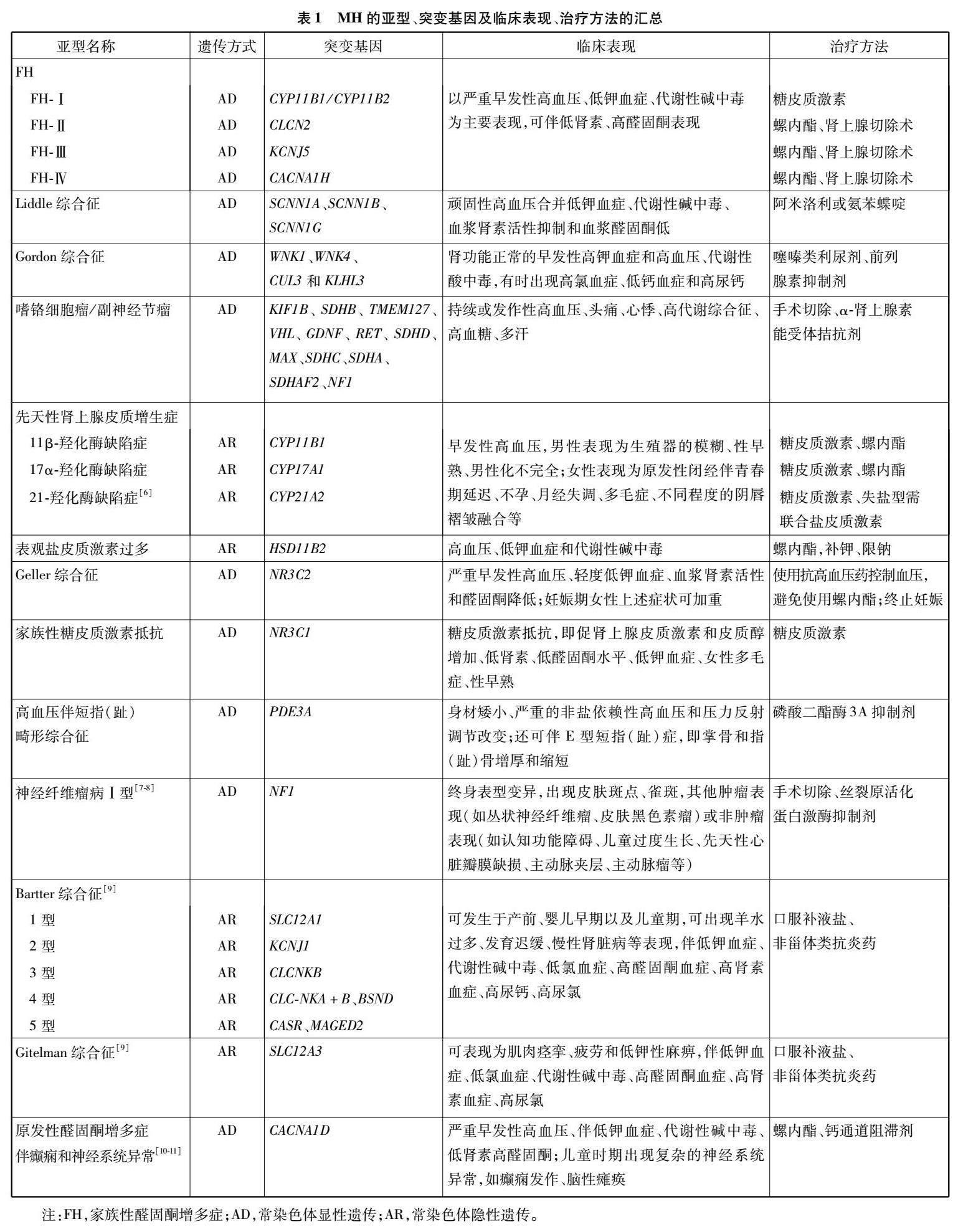
目前,已明确突变基因的MH亚型有22种(见表1)。在这里,我们详述了MH几种常见亚型及其遗传方式、突变基因、临床表现、治疗方法
2.1 家族性醛固酮增多症
家族性醛固酮增多症(familial hyperaldosteronism,FH)是一种罕见的常染色体显性遗传(autosomal dominant,AD)的MH。它是一组以严重早发性高血压、低钾血症、代谢性碱中毒和血浆醛固酮/肾素浓度比值(aldosterone to renin ratio,ARR)升高(ARR>20)为特征的疾病[12]。在临床上,FH的各种亚型很难与散发性原发性醛固酮增多症(primary aldosteronism,PA)区分开来。根据潜在的遗传缺陷,FH可分为Ⅰ~Ⅳ型。其中,FH-Ⅰ约占PA的0.6%[12],是由11β-羟化酶基因(11 β-hydroxylase,CYP11B1)和醛固酮合成酶基因(aldosterone synthase,CYP11B2)的不对称交叉产生嵌合基因所致,该基因在CYP11B1的5端具有促肾上腺皮质激素(adrenocortical hormone,ACTH)反应启动子区,在CYP11B2的3端具有醛固酮合成酶编码区,导致醛固酮的产生依赖于ACTH的表达[13],由于糖皮质激素对该疾病有显著的积极作用,它也被命名为糖皮质激素可治性醛固酮增多症(glucocorticoid-remediable aldosteronism,GRA)。FH-Ⅱ在PA患者中的患病率为1.5%~6.0%[12],它是由CLCN2基因突变所致,CLCN2编码肾上腺肾小球中表达的电压门控氯离子通道2 型,该基因突变使肾小球细胞膜容易去极化并激活电压门控钙通道,上调编码醛固酮合成酶的CYP11B2的表达,从而使盐皮质激素通道过度激活[14]。FH-Ⅲ约占PA的0.3%[15],可能是由KCNJ5的突变引起的,KCNJ5编码G蛋白偶联内向整流钾通道4(G-protein-coupled inward rectifier K+ channels 4, GIRK4)该基因突变影响GIRK4的选择性,导致肾上腺皮质细胞中钾选择性的丧失,增强钠电导,增加钠的流入和膜的去极化,最终导致CYP11B2的表达升高[16]。FH-Ⅳ是由CACNA1H突变引起[17],在全球范围内的发病率尚不清楚。CACNA1H在肾上腺球状带中大量表达,并编码T型钙通道的α亚基(CaV3.2)。CaV3.2的异常激活,增加钙内流,引起去极化,导致醛固酮合成异常[18]。使用低剂量的皮质类固醇可以有效抑制GRA患者ACTH的分泌,进而降低醛固酮水平。而FH-Ⅱ、FH-Ⅲ、FH-Ⅳ均需要通过单侧肾上腺切除术联合盐皮质激素拮抗剂来缓解症状[19]。
2,2 Liddle综合征
Liddle综合征(liddle syndrome,LS)是一种常染色体显性遗传疾病,是由编码上皮钠通道(epithelial sodium channel,ENaC)a、b、c亚基的SCNN1A、SCNN1B和SCNN1G基因突变引起的。远端小管中过度活化的ENaC直接导致钠重吸收增加,继而导致血容量扩张[10,19]。它可能是MH最常见的形式之一。有研究[20]发现在中国年轻高血压患者中,LS发病率为0.91%~1.52%。然而,它在整个高血压人群中的患病率仍然未知。LS最具特征的表现是顽固性高血压合并低钾血症、代谢性碱中毒、血浆肾素活性抑制和血浆醛固酮低,通常与猝死和早发性高血压的阳性家族史有关。截至目前,全球共发现41种导致LS的突变体,其中22种由中国学者首次报道[19]。在LS的治疗中,首选ENaC阻断剂(阿米洛利和氨苯蝶啶),并辅助低钠饮食。在中国阜外医院的国家心血管疾病中心建立的世界上最大的LS随访队列中,共有来自13个家系的74名患者接受了阿米洛利治疗并定期随访,治疗有效率为90%[5,19]。
2.3 Gordon综合征
Gordon综合征(gordon syndrome,GS),又称2型假性低醛固酮减少症(pseudohypoaldosteronism type II,PHAⅡ),是由调节肾远曲小管Na-Cl协同转运蛋白(NaCl cotransporter,NCC)活性的WNK1、WNK4、CUL3和KLHL3的功能获得突变引起的,表现为肾功能正常的早发性高钾血症和高血压、代谢性酸中毒,有时出现高氯血症、低钙血症和高尿钙[10,19]。高钾血症是GS的一个特征,其值可能为9 mmol/L。然而,由于异质性突变,患者可以表现出正常的血钾水平。在治疗中,噻嗪类药物对NCC具有抑制作用,因此临床建议使用噻嗪类利尿剂,有助于改善电解质异常并使血压正常化。对于轻度变异的患者,单独限制饮食中的盐可能是有效的[5,19]。
2.4 嗜铬细胞瘤/神经节旁瘤
嗜铬细胞瘤/神经节旁瘤(pheochromocytoma/paraganglioma,PCC/PGL)也是MH的常见病因之一,在高血压患者中的患病率为0.1%~0.6%[21]。遗传性PCC可单独发生或作为某些遗传性肿瘤综合征的局部表现,如多发性内分泌肿瘤、神经纤维瘤病等[22]。携带易感基因的患者很有可能在年轻时发展为PCC/PGL[23]。迄今为止,已经报道了以下PCC/PGL易感基因:KIF1B、SDHB、TMEM127、VHL、GDNF、RET、SDHD、MAX、SDHC、SDHA、SDHAF2、NF1[24]。这些基因突变激活儿茶酚胺/交感神经系统导致儿茶酚胺的过度释放、浓度升高,在临床上可表现为持续或发作性高血压、头痛、心悸、高代谢状态、高血糖和多汗。在明确诊断后,功能性PCC通常通过手术切除,并在围手术期服用α肾上腺素能拮抗剂[19,25-26]。
2.5 先天性肾上腺皮质增生症
先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是一组CYP11B1和CYP17A1突变与糖皮质激素合成有关的疾病,包括11β-羟化酶缺乏症(11β-hydroxylase deficiency,11β-OHD)——Ⅳ型和17α-羟化酶缺乏症——Ⅴ型。该类突变导致具有盐皮质激素活性的肾上腺类固醇如脱氧皮质酮(deoxycorticosterone,DOC)的积聚,从而出现高血压。11 β-OHD占所有CAH的0,2%~8.0%[19]。由于这种疾病往往伴随着高雄激素血症,它会促进体细胞的快速生长和骨骼成熟。此外,还可以观察到女性的男性化和男性的早熟[10,19]。由于皮质醇合成能力下降和性激素产生受损,在CAH的Ⅴ型中可以观察到男性的男性化不完全或生殖器模糊,以及女性的原发性闭经伴青春期延迟[27]。CAH患者建议使用糖皮质激素与盐皮质激素受体(mineralocorticoid receptor,MR)拮抗劑螺内酯联合抑制ACTH[10,19]。
2.6 表观盐皮质激素过多
表观盐皮质激素过多(apparent mineralocorticoid excess,AME)是一种罕见的常染色体隐性遗传疾病,其发病率在整个高血压人群尚不清楚。该疾病由11β羟基类固醇脱氢酶2型(11β-hydroxysteroid dehydrogenase type II,11βHSD2)缺乏引起。在生理状态下,11βHSD2将皮质醇转化为可的松,从而保护MR免受皮质醇激活[28]。病理状态下,MR的激活可诱导高血压、低钾血症和代谢性碱中毒。在50%~75%的患者中,可以观察到由高尿钙引起的肾钙沉着症,更罕见的是由于慢性低钾血症引起的肾囊肿[14]。AME的诊断依靠测量24 h尿游离皮质醇与可的松的比值,正常比值为1:1,而AME患者的比值范围为6.7~33.0。AME患者建议服用MR拮抗剂螺内酯,辅以低钠饮食及低剂量地塞米松来抑制促肾上腺皮质激素而引起的高血压和碱中毒[19,29]。
2.7 Geller综合征
Geller综合征,又称妊娠加重高血压,具有AD遗传模式,由MR突变引起,该突变存在于染色体4q31上,在密码子810(S810L;MRL810)处亮氨酸取代丝氨酸。这一突变改变ENaC和钠钾泵结构域的构象[27],增加其表达和活性,类固醇激素(如孕酮)对MR的亲和力增加,导致肾脏钠重吸收和钾分泌,临床表现为严重早发性高血压、轻度低钾血症、血浆肾素活性降低和醛固酮低的妊娠加重的高血压[14]。Geller综合征的治疗方法包括使用噻嗪类利尿剂、ENaC拮抗剂和辅以低钠饮食。终止妊娠是使血压正常化的有效治疗方法[19]。
2.8 家族性糖皮质激素抵抗
家族性糖皮质激素抵抗也是一种罕见的综合征,是由糖皮质激素受体上NR3C1基因的失活突变引起的。该突变导致糖皮质激素受体对皮质醇无反应[10,19],表现为糖皮质激素抵抗,即血浆ACTH和皮质醇增加、醛固酮水平低、低肾素、低钾血症、女性多毛症、性早熟等。低剂量地塞米松治疗可抑制下丘脑-垂体-肾上腺轴的活动,改善盐皮质激素过多症、皮质醇增多症和高雄激素血症。MR拮抗剂的使用,如螺内酯和依普利酮,有助于控制家族性糖皮质激素抵抗中的高血压[10,19]。
2.9 高血压伴短指(趾)畸形综合征
高血压伴短指(趾)畸形综合征(hypertension and brachydactyly syndrome,HTNB)是一种罕见的常染色体显性疾病,是由磷酸二酯酶3A(phosphodiesterase 3A,PDE3A)基因功能获得突变引起的。该突变致使PDE3A酶活性增强,降解细胞内环磷酸腺苷或环磷酸鸟苷,加速血管平滑肌细胞增殖并导致高血压[10]。临床表现为身材矮小、脑血管异常、严重的非盐依赖性高血压。HTNB还可伴E型短指(趾)症,其特征是掌骨和指骨增厚和缩短。HTNB也与延髓头端中央外侧的神经血管接触有关,并可能导致压力失调[27]。这种疾病在青春期前的儿童中很难诊断。如果不及时治疗,患者50岁之前通常会发生卒中导致的死亡。使用β受体阻滞剂、α受体阻滞剂、钙通道阻滞剂或血管紧张素转化酶抑制剂/血管紧张素Ⅱ受体阻滞剂联合或单药治疗可显著降低高血压,也可使用潜在具有PDE3A抑制作用的药物[19,30]。
3 MH的诊断
目前MH尚无统一的诊断标准,主要依靠基因检测的诊断(诊断流程可见 图1)。为了提高基因检测结果解读的准确性,美国医学遗传学与基因组学会(The American College of Medical Genetics and Genomics,ACMG)编制了ACMG联合共识作为序列变异解释的标准和指南。

注:簡化MDRD公式计算,GFR=186×Scr﹣1.154×Age﹣0.203×0.742(女性)[GFR为肾小球滤过率,Scr为血清肌酐(mg/dL),Age为年龄(岁)][31-33]。
图1 MH诊断流程图
3.1 基因检测的方法
随着新一代测序技术的发展,越来越多的基因检测方法比如Panel测序、全外显子组测序(whole exome sequencing,WES)、全基因组测序(whole genome sequencing,WGS)等被应用于临床。其中,WES可以捕获所有的外显子区域,包含了>85%的致病突变(包括非编码区突变),适用于具有血缘关系或者具有严重症状,以及具有多个症状的患者,能显著提高诊断率。但WES也存在特殊基因组区域(如:重复序列、假基因等)难以捕获、特殊变异类型(复杂拷贝数变异、结构异常)难以识别等局限性[34]。而WGS可以对整个基因组检测,检测范围全面,但是存在测序深度低、突变分析准确度低、对新变异结果的解读困难等局限性[35]。因此需要临床医生选择合适的测序方法,目前MH的诊断推荐采用诊断性基因组测序或WES,并在相应的家族成员中应用Sanger测序来确认诊断[19](见表2)。
3,2 ACMG指南的应用
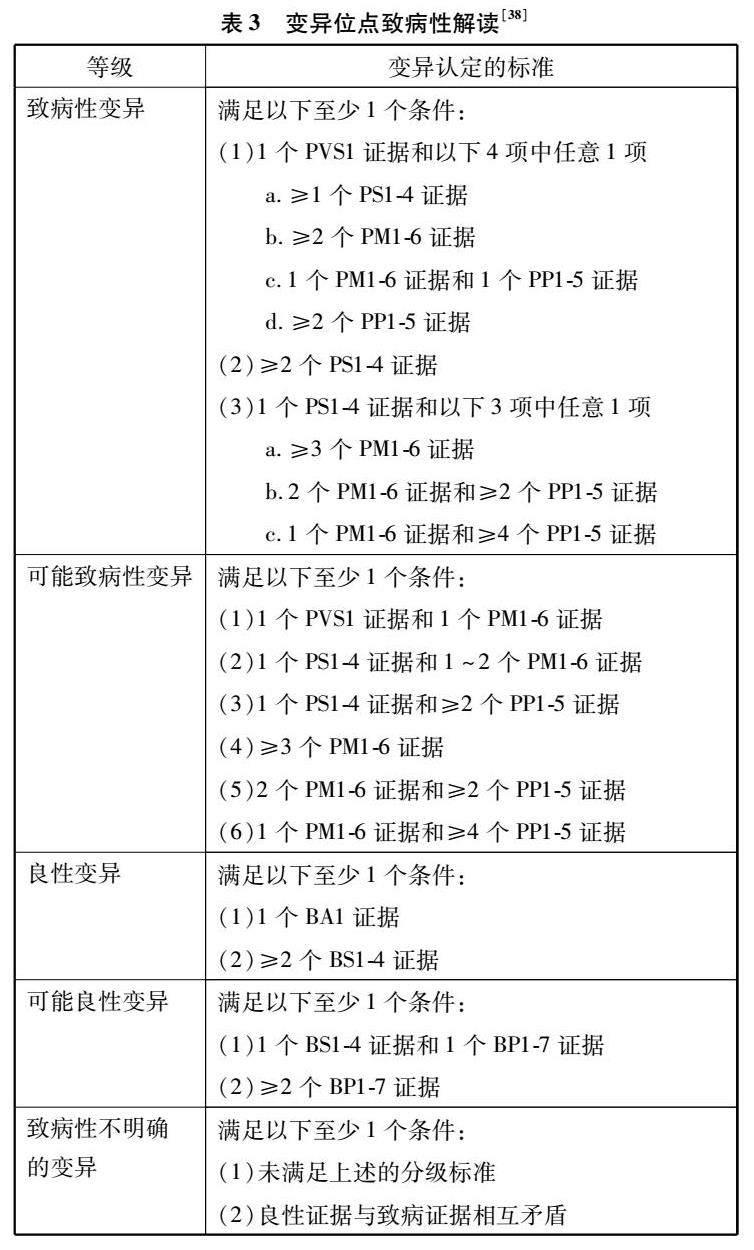
ACMG指南规范了MH诊断的流程和序列变体的解释标准,包括标本的选取、基因变异的分级和变异分类。首先,临床医生根据预期用途选择送检样本。若是产前可选择羊水和绒毛细胞等;若是产后进行体质评估可选择外周血,进行肿瘤性质评估可选用骨髓、外周血以及新鲜或冷冻的肿瘤组织等[36]。然后,检测结果的解读参照ACMG提供的两套标准:一套用于致病性或可能致病性变异,对变异程度分为非常强(PVS1)、强(PS1-4)、中等(PM1-6)或辅助证据(PP1-5);另一套用于良性或可能良性变异,对变异程度分为独立(BA1)、强(BS1-4)或辅助证据(BP1-6)。再根据组合分类标准将序列变体分为致病变异、可能致病变异、临床未明变异、可能良性变异以及良性变异,其致病性呈逐级降低(见表3)。其中临床未明变异又可分为临床意义未明1~5级,这更需临床医生结合患者及其家族史、其他实验室检查结果等全面的临床信息来评估变异。对于疑似隐性或新发变异,可将患者及其父母的样本一起纳入外显子组测序中进行分析[37],这不仅可以增强风险评估,以最大限度地减少过度诊断,同时还可促进对真正疾病患者的适当诊断。最后,为患者制定个性化的治疗方案,并定期跟踪随访治疗患者的血压和生化指标,以监测长期疗效并指导优化临床方案。
4 MH的治疗
MH患者暂无特异的治疗,通常采用对症治疗(见表1)。由于MH遗传模式的特异性以及发病机制的一致性,传统的对症降压治疗可能控制效果不佳。因此,在联合使用传统药物的基础上,开发针对特定基因位点的靶向药物的精准治疗成为高血压治疗的研究热点,并且已经取得了显著的成效。
反义寡核苷酸药物IONIS-AGT-LRx,作用靶点是肝脏合成的血管紧张素原(angiotensinogen,AGT),可抑制AGT的合成,降低血管紧张素Ⅱ的水平。该药物的Ⅱ期临床试验结果显示,在基础治疗的背景上,能有效降低收缩压和舒张压[38]。小干扰RNA药物zilebesiran,是一种化学修饰偶联N-乙酰半乳糖胺的小干扰RNA,以AGT为靶点,抑制血管紧张素Ⅰ和Ⅱ的产生,从而达到降压效果,通过每隔6个月皮下给药的治疗方案,也有可能提供比现有治疗更明显的优势[39]。醛固酮合成酶抑制剂Baxdrostat,可抑制醛固酮的合成,从而降低血压[40]。与此同时,血管紧张素1-7类似物、血管紧张素Ⅱ导向疫苗、奈普赖氨酸抑制剂、内皮素受体拮抗剂、多巴胺β-羟化酶抑制剂和脑氨肽酶A抑制剂等药物正处于临床开发的不同阶段[41]。此外,经皮肾动脉交感神经消融术通过降低肾动脉交感神经活性,阻断交感神经过度兴奋,使血压下降,也可以成为一种新的治疗选择[41]。动物实验中使用CRISPR-Cas9基因编辑技术靶向介导肝脏AGT,可以持续降低血压,提示此技术在将来可成为人类持续甚至终身控制高血压的潜在疗法[42]。
5 總结
MH的具体发病机制尚不能确定,临床辨识困难且无统一的诊治标准。现综述了在此疾病中常见亚型的基因机制,强调了该病进行基因诊断的必要性,以及结合ACMG指南进行解读分析的重要性。由于此病的复杂性和异质性,对相关患者群体进行遗传学检测和分析,将有助于更深层次地认识该疾病。
参 考 文 献
Whelton PK,Flack JM,Jennings GLR,et al. Editors' commentary on the 2023 ESH management of arterial hypertension guidelines[J]. Hypertension,2023,80(9):1795-1799.
Lu YT,Fan P,Zhang D,et al. Overview of monogenic forms of hypertension combined with hypokalemia[J]. Front Pediatr,2021,8:543309.
Raina R,Krishnappa V,Das A,et al. Overview of monogenic or Mendelian forms of hypertension[J]. Front Pediatr,2019,7:263.
Park SJ,Shin JI. Diagnosis and treatment of monogenic hypertension in children[J]. Yonsei Med J,2023,64(2):77-86.
Khandelwal P,Deinum J. Monogenic forms of low-renin hypertension:clinical and molecular insights[J]. Pediatr Nephrol,2022,37(7):1495-1509.
Travers S,Bouvattier C,Fagart J,et al. Interaction between accumulated 21-deoxysteroids and mineralocorticoid signaling in 21-hydroxylase deficiency[J]. Am J Physiol Endocrinol Metab,2020,318(2):E102-E110.
Wang W,Wei CJ,Cui XW,et al. Impacts of NF1 gene mutations and genetic modifiers in neurofibromatosis type 1[J]. 2021,12:704639.
de Blank PMK,Gross AM,Akshintala S,et al. MEK inhibitors for neurofibromatosis type 1 manifestations:clinical evidence and consensus[J]. Neuro Oncol,2022,24(11):1845-1856.
Nu?ez-Gonzalez L,Carrera N,Garcia-Gonzalez MA. Molecular basis,diagnostic challenges and therapeutic approaches of Bartter and Gitelman syndromes:a primer for clinicians[J]. Int J Mol Sci,2021,22(21):11414.
Ostrowska A,Skrzypczyk P. Monogenic hypertension[J]. Pol Merkur Lekarski,2022,50(297):198-201.
Semenova NA,Ryzhkova OR,Strokova TV,et al.Treti? slucha? sindroma pervichnogo al'dosteronizma, sudorog i nevrologicheskikh narusheni? (PASNA), obuslovlennogo variantom mutatsii de novo v gene CACNA1D[The third case report a patient with primary aldosteronism,seizures,and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene][J]. Zh Nevrol Psikhiatr Im S S Korsakova,2018,118(12):49-52.
Araujo-Castro M,Martín Rojas-Marcos P,Parra Ramírez P. Familial forms and molecular profile of primary hyperaldosteronism[J]. Hipertens Riesgo Vasc,2022,39(4):167-173.
Funder JW,Carey RM,Mantero F,et al. The Management of Primary Aldosteronism:Case Detection,Diagnosis,and Treatment:An Endocrine Society Clinical Practice Guideline[J]. J Clin Endocrinol Metab,2016,101(5):1889-1916.
Scholl UI,St?lting G,Schewe J,et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II[J]. Nat Genet,2018,50(3):349-354.
Mulatero P,Tauber P,Zennaro MC,et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism[J]. Hypertension,2012,59(2):235-240.
Monticone S,Hattangady NG,Nishimoto K,et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells[J]. J Clin Endocrinol Metab,2012,97(8):E1567-E1572.
Scholl UI,St?lting G,Nelson-Williams C,et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism[J]. Elife,2015,4:e06315.
Daniil G,Fernandes-Rosa FL,Chemin J,et al. CACNA1H mutations are associated with different forms of primary aldosteronism[J]. EBioMedicine,2016,13:225-236.
Lu Y,Fan P,Hakonarson H,et al. Monogenic hypertension-a type of "curable" hypertension[J]. Sci Bull (Beijing),2023,68(7):657-660.
Tetti M,Monticone S,Burrello J,et al. Liddle syndrome:review of the literature and description of a new case[J]. Int J Mol Sci,2018,19(3):812.
Kavinga Gunawardane PT,Grossman A. The clinical genetics of phaeochromocytoma and paraganglioma[J]. Arch Endocrinol Metab,2017,61(5):490-500.
Tsirlin A,Oo Y,Sharma R,et al. Pheochromocytoma:a review[J]. Maturitas,2014,77(3):229-38.
N?lting S,Ullrich M,Pietzsch J,et al. Current management of pheochromocytoma/paraganglioma:a guide for the practicing clinician in the era of precision medicine[J]. Cancers (Basel),2019,11(10):1505.
Sbardella E,Cranston T,Isidori AM,et al. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas[J]. Endocrine,2018,59(1):175-182.
Patel D,Phay JE,Yen TWF,et al. Update on pheochromocytoma and paraganglioma from the SSO endocrine and head and neck disease site working group,part 2 of 2:perioperative management and outcomes of pheochromocytoma and paraganglioma[J]. Ann Surg Oncol,2020,27(5):1338-1347.
Lenders JWM,Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma[J]. Endocrinol Metab (Seoul),2017,32(2):152-161.
Levanovich PE,Diaczok A,Rossi NF. Clinical and molecular perspectives of monogenic hypertension[J]. Curr Hypertens Rev,2020,16(2):91-107.
Bulsari K,Falhammar H.Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency[J]. Endocrine,2017,55(1):19-36.
Aggarwal A,Rodriguez-Buritica D. Monogenic hypertension in children:a review with emphasis on genetics[J]. Adv Chronic Kidney Dis,2017,24(6):372-379.
Toka O,Tank J,Sch?chterle C,et al. Clinical effects of phosphodiesterase 3A mutations in inherited hypertension with brachydactyly[J]. Hypertension,2015,66(4):800-808.
Levey AS,Bosch JP,Lewis JB,et al. A more accurate method to estimate glomerular filtration rate from serum creatinine:a new prediction equation. Modification of Diet in Renal Disease Study Group[J]. Ann Intern Med,1999,130(6):461-470.
Rule AD,Larson TS,Bergstralh EJ,et al. Using serum creatinine to estimate glomerular filtration rate:accuracy in good health and in chronic kidney disease[J]. Ann Intern Med,2004,141(12):929-937.
Levey AS,Coresh J,Greene T,et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate[J]. Ann Intern Med,2006,145(4):247-254.
Rexach J,Lee H,Martinez-Agosto JA,et al. Clinical application of next-generation sequencing to the practice of neurology[J]. Lancet Neurol,2019,18(5):492-503.
Wu S,Schmitz U. Single-cell and long-read sequencing to enhance modelling of splicing and cell-fate determination[J]. Comput Struct Biotechnol J,2023,21:2373-2380.
Shao L,Akkari Y,Cooley LD,et al. Chromosomal microarray analysis,including constitutional and neoplastic disease applications,2021 revision:a technical standard of the American College of Medical Genetics and Genomics (ACMG)[J]. Genet Med,2021,23(10):1818-1829.
Richards S,Aziz N,Bale S,et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med,2015,17(5):405-424.
Morgan ES,Tami Y,Hu K,et al. Antisense inhibition of angiotensinogen with IONIS-AGT-LRx:results of phase 1 and phase 2 studies[J]. JACC Basic Transl Sci,2021,6(6):485-496.
Desai AS, Webb DJ, Taubel J, et al. Zilebesiran, an RNA Interference Therapeutic Agent for Hypertension[J]. N Engl J Med. 2023,389(3):228-238.
Freeman MW,Halvorsen YD,Marshall W,et al. Phase 2 trial of baxdrostat for treatment-resistant hypertension[J]. N Engl J Med,2023,388(5):395-405.
Ranasinghe P,Addison ML,Webb DJ. Small interfering RNA therapeutics in hypertension:a viewpoint on vasopressor and vasopressor-sparing strategies for counteracting blood pressure lowering by angiotensinogen-targeting small interfering RNA[J]. J Am Heart Assoc,2022,11(20): e027694.
Sun H,Hodgkinson CP,Pratt RE,et al. CRISPR/Cas9 mediated deletion of the angiotensinogen gene reduces hypertension:a potential for cure?[J]. Hypertension,2021,77(6):1990-2000.
收稿日期:2023-06-08
