
HTRA3基因对脉络膜新生血管和M2型巨噬细胞极化的影响△
2024-04-18 00:33:52肇莉莉孙连义马为梅
眼科新进展 2024年4期
肇莉莉 王 萍 孙连义 马为梅 张 乐 喻 磊
湿性年龄相关性黄斑变性(wAMD)是一种累及视网膜黄斑区的眼病之一,在老年人群中具有较高的发病率[1-2]。脉络膜新生血管(CNV)是wAMD的典型病变,也是导致患者视力丧失的主要原因[3]。目前,虽然玻璃体内注射血管内皮生长因子(VEGF)抑制剂在治疗wAMD方面取得了一定疗效[4],然而,抗VEGF药物存在耐药性问题[5],并且会引起眼压升高[6]、眼部感染[7]等并发症,因此,仍需要开发AMD的新型治疗手段。免疫微环境失衡是导致wAMD的始发因素,wAMD病变部位出现大量巨噬细胞浸润,这些巨噬细胞通过分泌VEGF和炎性因子促进血管内皮细胞增殖并引起新生血管渗漏,导致wAMD形成[8]。巨噬细胞具有可塑性,可极化为M1和M2型,其中M2型极化可导致脉络膜血管形成[9-10]。因此,调控巨噬细胞表型是治疗wAMD的有效途径。HtrA丝氨酸肽酶3(HTRA3)是2003年首次报道的一个蛋白质编码基因,位于染色体4p16.1上[11]。HTRA3与HTRA1类似,都能抑制转化生长因子-β家族蛋白介导的信号通路[12]。据报道,HTRA1基因启动子里的一个单核苷酸多态性与AMD有关联,含有HTRA1多态性的人群发生AMD的危险度比正常人高10倍[13-14]。HTRA3在AMD小鼠视网膜中表达升高,提示HTRA3很可能参与AMD病变的进展[15]。另外,HTRA3可能调节免疫细胞功能[16]。本研究旨在揭示HTRA3基因对CNV和M2型巨噬细胞极化的影响,从而开发AMD的新型治疗靶标。
1 材料与方法
1.1 材料
1.1.1 实验试剂
慢病毒载体由吉玛基因合成。RPMI 1640培养基(货号:22400105)购自美国Gibco公司。氯化钴(CoCl2)(货号:C8661)购自美国Sigma公司。Lipofectamine2000(货号:11668027)购自美国Invitrogen公司。Matrigel(货号:356234)购自美国BD Biosciences公司。苏木精伊红(HE)染色试剂盒(货号:C0105S)、TRIzol(货号:R0016)、RIPA裂解液(货号:P0013B)、ECL试剂盒(货号:P0018S)购自碧云天生物技术研究所。荧光素钠(货号:A833)购自美国Thermo Fisher公司。Prime Script RT Master Mix试剂盒(货号:RR036A)购自日本TaKaRa公司。SYBR Green Master Mix(货号:4367659)购自美国ABI公司。HTRA3(货号:ab227463)、VEGFA(货号:ab46154)、核因子κB(NF-κB)p65(货号:ab288751)、Lamin B(货号:ab32535)和β-actin(货号:ab8226)一抗以及HRP标记的IgG二抗(货号:ab205718)购自英国Abcam公司。
1.1.2 实验细胞和动物
恒河猴脉络膜血管内皮细胞系(RF/6A)购自美国ATCC。60只SPF级雄性老龄C57BL/6J小鼠(18~20个月龄,体重35~40 g)由西北大学提供[SYXK(陕)2021-004]。小鼠在25 ℃、55%相对湿度、12 h循环照明环境中使用标准小鼠饲料和纯净水饲养。
1.2 方法
1.2.1 血清样本收集
2019年1月至2022年8月期间,收集西安市人民医院(西安市第四医院)就诊的30例wAMD患者为wAMD组,其中男16例,女14例,年龄51~78(68.81±6.09)岁,参考wAMD国际诊断标准[17]对患者进行诊断。剔除伴有其他视网膜疾病、恶性肿瘤、心脑血管系统疾病、器官病变及接受治疗的wAMD患者。收集同期来院体检的健康者30例为健康组,其中男15例,女15例,年龄53~81(70.43±6.42)岁。健康组与wAMD组受试者的年龄、性别比例相比,差异均无统计学意义(均为P>0.05)。收集两组受试者的空腹静脉血,qRT-PCR检测血清HTRA3 mRNA水平。本研究通过西安市人民医院(西安市第四医院)伦理委员会审批(审批号:201901170025),受试者均知情同意。
1.2.2 细胞培养与转染处理
将RF/6A细胞培养在添加体积分数10%胎牛血清和10 g·L-1双抗的RPMI 1640培养基中,置于37 ℃和含体积分数5%CO2的细胞培养箱中培养。细胞生长至对数期后用于实验。将RF/6A细胞随机分为对照组、NC-sh组和HTRA3-sh组,6孔板中接种RF/6A细胞并培养至80%融合,按照试剂盒说明,使用Lipofectamine2000将NC-shRNA和HTRA3-shRNA慢病毒载体分别转染到NC-sh组和HTRA3-sh组RF/6A细胞。对照组细胞不进行转染。转染48 h后,通过qRT-PCR和Western blot检测HTRA3的转染效率。每组设置6个复孔。
1.2.3 细胞分组及处理
验证HTRA3转染效率后,将RF/6A细胞随机分为N组、H组、H+NC-sh组和H+HTRA3-sh组。其中,H+NC-sh组和H+HTRA3-sh组细胞分别转染NC-shRNA和HTRA3-shRNA;N组RF/6A细胞在完全RPMI 1640培养基中进行常氧培养,其他组细胞在添加200 mmol·L-1CoCl2的RPMI 1640培养基中进行低氧培养。每组设置6个复孔。
1.2.4 小管形成测定
使用Matrigel(每孔80 μL)包被96孔板,室温静置30 min,加入1.2.3分组处理后的细胞30 μL(每孔5×104个RF/6A细胞),培养6 h后在显微镜下观察小管形成情况,Image-Pro Plus 6.0软件计算闭合管腔数量。每组设置6个复孔。
1.2.5 激光诱导CNV小鼠模型
使用复方托吡酰胺滴眼液对小鼠进行散瞳,10 g·L-1苯巴比妥钠经腹腔注射麻醉小鼠(10 mL·kg-1)。围绕视盘用氩激光(激光波长350 nm,输出功率350 mW,曝光时间0.1 s,光斑直径75 μm)对小鼠双眼发射四个激光点。光凝后有白色气泡产生证实Bruch膜破裂,表明建模成功。
1.2.6 动物分组及处理
将小鼠随机分为对照组、CNV组、CNV+NC-sh组和CNV+HTRA3-sh组,每组12只。对照组为未建模的小鼠,其他组为CNV建模成功的小鼠。使用盐酸奥布卡因进行表面麻醉,然后使用30G穿刺针头向CNV+NC-sh组和CNV+HTRA3-sh组小鼠玻璃体内分别注射1 μL滴度为1×1011TU·mL-1的NC-shRNA和HTRA3-shRNA慢病毒载体,对照组和CNV组小鼠玻璃体内注射PBS,均为双眼注射。注射后给予左氧氟沙星滴眼液抗感染。
1.2.7 荧光素眼底血管造影
注射后7 d,所有小鼠均行荧光素眼底血管造影(FFA)检查。使用复方托吡酰胺滴眼液对各组小鼠进行散瞳,10 g·L-1苯巴比妥钠经腹腔注射麻醉小鼠(10 mL·kg-1)。然后经腹腔注射0.05 mL 100 g·L-1荧光素钠,2~3 min后拍摄眼底照片,在480 nm激发光和525 nm发射光下使用滤光片获取图像。使用ImageJ软件计算CNV的平均荧光强度。
1.2.8 样本收集
所有小鼠FFA检查结束后处死,摘取双眼眼球,取双眼中任意一只眼球行40 g·L-1多聚甲醛固定,用于HE染色;另一只眼球在显微镜下冰上小心去除结缔组织和肌肉,去除眼前节,分离出脉络膜组织,与生理盐水混合并研磨匀浆,用于qRT-PCR和Western blot检测。
1.2.9 眼球HE染色
40 g·L-1多聚甲醛固定眼球24 h。常规制成4 μm厚的石蜡切片,根据试剂盒说明进行HE染色。
1.2.10 qRT-PCR检测
通过qRT-PCR检测wAMD患者和健康体检者的血清中HTRA3 mRNA水平,并检测RF/6A细胞或各组小鼠脉络膜组织中HTRA3、类几丁质酶3样蛋白3(Ym-1)、精氨酸酶1(Arg-1)、诱导型一氧化氮合酶(iNOS)、环氧化酶-2(COX-2)和VEGFA mRNA水平。TRIzol试剂提取总RNA。使用逆转录试剂盒将RNA逆转录为cDNA,然后在ABI Prism 7900HT型荧光定量PCR仪上使用SYBR Green Master Mix进行扩增。扩增程序:95 ℃ 30 s,95 ℃ 5 s、60 ℃ 30 s,循环40次。引物序列见表1。GAPDH为内参基因。通过2-ΔΔCt法计算基因相对表达量。

表1 引物序列
1.2.11 Western blot检测
通过RIPA裂解液提取RF/6A细胞或小鼠脉络膜组织总蛋白,然后在100 g·L-1SDS-PAGE上分离蛋白并转移到PVDF膜上,50 g·L-1脱脂牛奶室温封闭30 min。然后与1:5 000稀释的HTRA3、VEGFA、NF-κB p65、Lamin B和β-actin一抗在4 ℃下孵育过夜。然后将膜与1:5 000稀释的HRP标记的IgG二抗在室温下孵育2 h。ECL显影,ImageJ软件测定条带灰度值。Lamin B作为核蛋白内参,β-actin 作为总蛋白内参。
1.3 统计学分析
使用SPSS 21.0软件行统计分析,定量数据采用均数±标准差表示。组间差异比较采用t检验或单向方差分析。检验水准:α=0.05。
2 结果
2.1 wAMD患者和健康体检者的血清HTRA3 mRNA水平
与健康组(1.00±0.33)比较,wAMD组患者的血清中HTRA3 mRNA水平(2.41±0.57)升高,差异有统计学意义(t=11.804,P<0.001)。
2.2 下调HTRA3抑制缺氧诱导的RF/6A细胞小管形成
RF/6A细胞转染后,与对照组和NC-sh组比较,HTRA3-sh组RF/6A细胞的HTRA3 mRNA和蛋白表达水平均降低(均为P<0.05)(表2和图1)。缺氧处理后,与N组比较,H组RF/6A细胞的闭合管腔数量、HTRA3和VEGFA的mRNA和蛋白表达水平均增加(均为P<0.05);与H+NC-sh组比较,H+HTRA3-sh组RF/6A细胞的闭合管腔数量、HTRA3和VEGFA的mRNA和蛋白表达水平均减少(均为P<0.05);H组与H+NC-sh组RF/6A细胞的闭合管腔数量、HTRA3和VEGFA的mRNA和蛋白表达水平相比,差异均无统计学意义(均为P>0.05)(表3、图2、表4和图3)。

图1 Western blot检测各组RF/6A细胞HTRA3蛋白表达
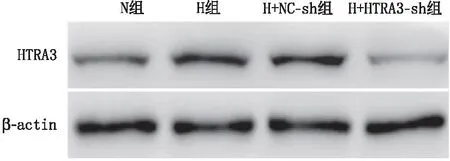
图2 Western blot检测各组RF/6A细胞HTRA3蛋白表达
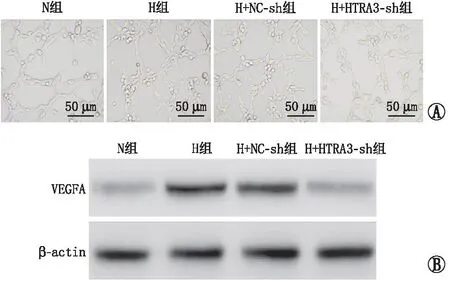
A:各组RF/6A细胞的小管形成图;B:各组RF/6A细胞的VEGFA蛋白条带。图3 下调HTRA3对缺氧诱导的RF/6A细胞小管形成和VEGFA蛋白表达的影响

表2 各组RF/6A细胞的HTRA3 mRNA和蛋白相对表达量

表3 各组RF/6A细胞HTRA3 mRNA和蛋白相对表达量
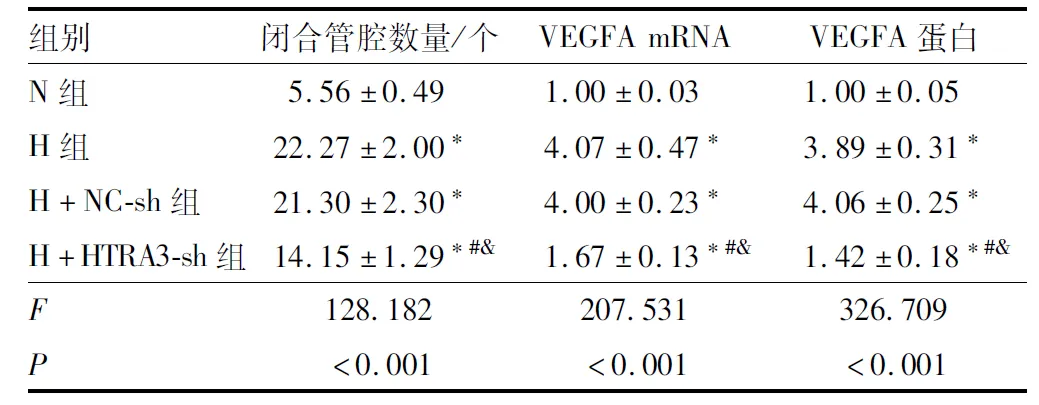
表4 各组RF/6A细胞的闭合管腔数量、VEGFA mRNA和蛋白相对表达量
2.3 下调HTRA3抑制CNV小鼠脉络膜血管生成
与对照组比较,CNV组小鼠的HTRA3 mRNA和蛋白表达水平均增加(均为P<0.05);与CNV+NC-sh组比较,CNV+HTRA3-sh组小鼠的HTRA3 mRNA和蛋白表达水平均减少(均为P<0.05);CNV组与CNV+NC-sh组小鼠的HTRA3 mRNA和蛋白表达水平相比,差异均无统计学意义(均为P>0.05)(表5和图4)。对照组小鼠脉络膜形态正常;CNV组和CNV+NC-sh组小鼠脉络膜结构紊乱,脉络膜增生,Bruch膜破裂,血管生成增加;CNV+HTRA3-sh组小鼠脉络膜病变减轻,脉络膜增生和血管生成减少(图5)。与对照组比较,CNV组小鼠的CNV相对荧光强度升高(P<0.05);与CNV+NC-sh组比较,CNV+HTRA3-sh组小鼠的CNV相对荧光强度降低(P<0.05);CNV组与CNV+NC-sh组小鼠的CNV相对荧光强度相比,差异无统计学意义(P>0.05)(表5和图5)。
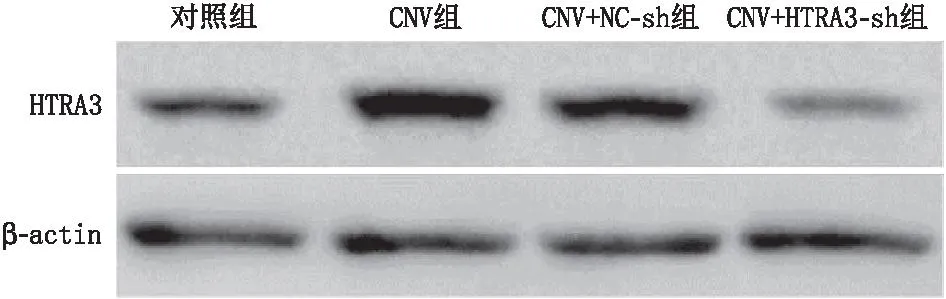
图4 Western blot检测各组小鼠脉络膜组织中HTRA3蛋白表达
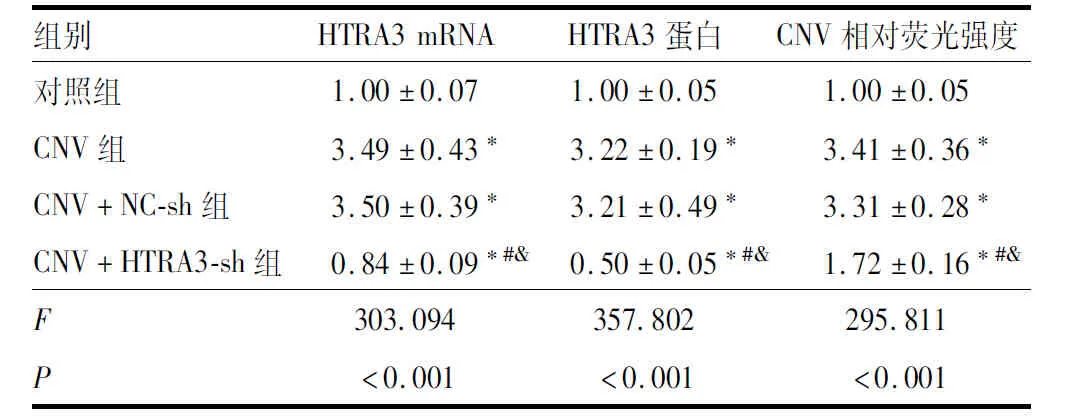
表5 各组小鼠脉络膜组织中的HTRA3 mRNA和蛋白相对表达量以及CNV相对荧光强度
2.4 下调HTRA3对CNV小鼠脉络膜组织中巨噬细胞极化的影响
与对照组比较,CNV组小鼠脉络膜组织中M2型巨噬细胞标志物Ym-1和Arg-1 mRNA水平升高,M1型巨噬细胞标志物iNOS和COX-2 mRNA水平降低(均为P<0.05);与CNV+NC-sh组比较,CNV+HTRA3-sh组小鼠脉络膜组织中Ym-1和Arg-1 mRNA水平降低,iNOS和COX-2 mRNA水平升高(均为P<0.05);CNV组与CNV+NC-sh组小鼠脉络膜组织中Ym-1、Arg-1、iNOS和COX-2 mRNA水平相比,差异均无统计学意义(均为P>0.05)(表6)。

表6 各组小鼠脉络膜组织中Ym-1、Arg-1、iNOS和COX-2的mRNA相对表达量
2.5 下调HTRA3对CNV小鼠脉络膜组织NF-κB信号通路的影响
对照组、CNV组、CNV+NC-sh组和CNV+HTRA3-sh组小鼠脉络膜组织中的细胞核NF-κB p65蛋白相对表达量依次为1.00±0.05、3.41±0.27、3.39±0.35、1.87±0.11(F=315.393,P<0.001)。与对照组比较,CNV组小鼠的脉络膜组织中细胞核NF-κB p65蛋白表达水平升高(P<0.05);与CNV+NC-sh组比较,CNV+HTRA3-sh组小鼠的脉络膜组织中细胞核NF-κB p65蛋白表达水平降低(P<0.05);CNV组与CNV+NC-sh组小鼠脉络膜组织中细胞核NF-κB p65蛋白表达水平相比,差异无统计学意义(P>0.05)(图6)。

图6 Western blot检测各组小鼠脉络膜组织中的细胞核NF-κB p65蛋白表达
3 讨论
CNV是wAMD患者视力丧失的主要原因,虽然抗VEGF治疗可以抑制CNV形成,但也可能引起多种并发症[6-7],因此,仍需要开发新型wAMD治疗手段和治疗靶点。HtrA家族是一种热休克诱导的蛋白酶家族,在进化过程中非常保守,该家族成员不仅参与调节细胞存活、线粒体稳态、胎盘发育等多种生理功能,而且参与癌症、神经退行性疾病、关节炎等多种病理过程的发生和发展[18]。目前,HtrA家族成员中HTRA1的研究较多。据报道,HTRA1多态性与AMD的发生相关[13-14]。CNV患者房水中的HTRA1水平高于白内障患者,并且经雷珠单抗玻璃体内注射治疗后HTRA1水平降低[19]。HTRA-1调节血管生成功能,HTRA-1的升高与早产儿视网膜病变的发生风险相关[20]。然而,HTRA3在wAMD中的研究较少。本研究观察到wAMD患者的血清HTRA3 mRNA水平较健康人明显升高,提示HTRA3可能参与了AMD的发病过程。由于HTRA3与HTRA1具有高度同源性和结构相似性,这两种酶在人体组织中的表达模式相似,提示它们可能具有相似的生物学活性和互补的生理功能[12]。据报道,HTRA3在AMD小鼠视网膜中表达升高[15]。
视网膜缺氧是wAMD的主要病因之一[21],缺氧可诱导CNV形成[22-23]。本研究通过转染HTRA3-shRNA慢病毒下调了RF/6A细胞中的HTRA3,然后对RF/6A细胞进行缺氧处理,结果表明,下调HTRA3抑制了缺氧诱导的RF/6A细胞小管形成,并下调了VEGFA的表达。另外,激光诱导CNV是一种模拟了人类wAMD的动物建模方法[24]。本研究建立了激光诱导的CNV小鼠模型,然后对小鼠玻璃体内注射HTRA3-shRNA慢病毒,结果表明,下调HTRA3减轻了CNV小鼠脉络膜病变,抑制了血管生成和CNV生成。Nie等[25]报道,HTRA3是一种含有胰岛素样生长因子(IGF)结合结构域的新型丝氨酸蛋白酶,在小鼠胎盘形成过程中在母胎界面选择性表达,HTRA3在介导血管和胎盘形成中起着关键作用。本研究证实,HTRA3水平影响RF/6A细胞小管形成及血管生成因子VEGFA的表达水平。据报道,HTRA3是一种调节脑内皮细胞特异性应答的基因,与前脑血管发育有关[26]。敲除HTRA3抑制了滋养层细胞的侵袭和血管生成[27]。这些结果说明HTRA3可促进血管生成和CNV形成。
wAMD发病过程中免疫微环境失衡,大量巨噬细胞浸润是免疫微环境失衡的重要因素,因为巨噬细胞可促进血管内皮细胞增殖并引起新生血管渗漏[8]。wAMD发病过程中巨噬细胞主要通过M2型极化诱导CNV形成[9-10]。巨噬细胞发生M2型极化后可分泌包括VEGF在内的多种促血管生成因子。本研究结果表明,下调HTRA3降低了CNV小鼠脉络膜组织中M2型巨噬细胞标志物(Ym-1和Arg-1)的表达,上调了M1型巨噬细胞标志物(iNOS和COX-2)的表达,抑制了M2型巨噬细胞极化。有学者研究表明,HTRA家族的基因突变影响巨噬细胞的存活率[28]。另外,HTRA3与胃癌的免疫细胞浸润程度相关[16]。HTRA3的高表达与获得性免疫细胞(Th17细胞、T辅助细胞、T中央记忆细胞等)呈负相关,与天然免疫细胞(NK细胞、肿瘤相关巨噬细胞、未成熟树突状细胞等)呈正相关[16]。因此我们推测,HTRA3可能通过促进M2型巨噬细胞极化参与了wAMD发病过程。
NF-κB信号通路参与激活wAMD患者脉络膜和视网膜中的炎症反应[29],NF-κB信号通路的激活与CNV形成有关[30],NF-κB信号通路的激活在包括wAMD在内的多种疾病中起到促进血管生成作用[31-32]。因此,抑制NF-κB信号通路是治疗wAMD的有效策略[33]。其他学者报道,HTRA3高表达可能激活NF-κB通路[16]。本研究结果表明,下调HTRA3抑制CNV小鼠脉络膜组织NF-κB的核转位。据报道,NF-κB p65可直接与HTRA1启动子(氨基酸347)结合,阻断HTRA1可显著抑制NF-κB信号通路[34]。考虑到HTRA3与HTRA1具有高度同源性和结构相似性,并且表达模式相似[12],我们推测,HTRA3可能与NF-κB p65具有结合位点,下调HTRA3对CNV形成的抑制作用也可能与抑制NF-κB信号通路有关。
4 结论
HTRA3可促进血管生成和CNV形成,可能通过促进M2型巨噬细胞极化参与了wAMD的发病过程,通过下调HTRA3可抑制CNV形成和M2型巨噬细胞极化,该作用可能与NF-κB信号通路有关。本研究结果提示,HTRA3可能是防治wAMD的重要潜在靶点。
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01 06:08:32
服饰导报·鞋世界(2021年4期)2021-05-17 14:01:41
中医眼耳鼻喉杂志(2019年3期)2019-04-13 05:26:46
中医眼耳鼻喉杂志(2019年3期)2019-04-13 05:26:30
中国药理学通报(2019年5期)2019-01-11 18:03:39
大连医科大学学报(2018年4期)2018-04-11 04:00:53
电子测试(2017年15期)2017-12-18 07:18:51
电源技术(2015年1期)2015-08-22 11:16:18
测绘科学与工程(2014年6期)2014-02-27 07:06:21
湖南中医药大学学报(2013年9期)2013-03-11 16:33:43
