
基因组和DNA甲基化组联合分析筛选猪肉质性状关键基因
2024-04-11 07:28:34赵真坚王凯陈栋申琦余杨崔晟頔王俊戈陈子旸禹世欣陈佳苗王翔枫唐国庆
中国农业科学 2024年7期
赵真坚,王凯,陈栋,申琦,余杨,崔晟頔,王俊戈,陈子旸,禹世欣,陈佳苗,王翔枫,唐国庆
基因组和DNA甲基化组联合分析筛选猪肉质性状关键基因
赵真坚,王凯,陈栋,申琦,余杨,崔晟頔,王俊戈,陈子旸,禹世欣,陈佳苗,王翔枫,唐国庆
四川农业大学动物科技学院/农业农村部畜禽生物组学重点实验室/畜禽遗传资源发掘与创新利用四川省重点实验室/猪禽种业全国重点实验室,成都 611130
【背景】猪的肉质性状是重要的经济性状。研究影响各类肉质性状的分子机制,发掘关键基因,从而指导猪的遗传改良,对改善猪肉品质具有重要意义。当前肉质相关的机制研究主要是基于DNA的基因组研究,而对肉质性状的DNA甲基化研究以及结合基因组和甲基化组的综合分析却鲜有报道。【目的】通过基因组和DNA甲基化组联合分析,筛选、鉴定了影响猪肉质的潜在关键基因。为猪肉品质的遗传改良研究提供借鉴。【方法】检测了140头大白猪背最长肌的28个肉质性状,通过表观基因组关联分析(EWAS)与全基因组关联分析(GWAS)筛选了各性状显著关联的CpG和SNP位点。随后以SNP为协变量对GWAS和EWAS重叠的显著关联位点进行条件关联分析,进一步筛选具有独立效应的CpG位点。然后以CpG位点的甲基化水平为因变量,以SNP为自变量进行关联分析从而鉴定甲基化数量性状位点(meQTL)。最后使用顺式甲基化数量性状位点(cis-meQTL)作为工具变量进行孟德尔随机化分析,从而推断cis-meQTL与表型之间的因果关系,同时对位点进行注释,鉴定潜在的关键基因。【结果】(1)在屠宰45 min黄度值(b45min)、滴水损失(DL)、二十二碳六烯酸(C22:6n-3)3个肉质性状上,EWAS和GWAS在相同基因组区域鉴定到显著关联位点。(2)b45min的7个CpG位点在条件关联分析后仍保持显著,DL有1个CpG位点在条件关联分析后仍保持显著,而C22:6n-3的3个位点在使用SNP作为协变量分析后不再显著,表明EWAS鉴定的b45min的7个CpG位点和DL的1个CpG位点的显著关联不受附近的显著SNP影响。(3)b45min的7个CpG位点和DL的1个CpG位点共鉴定了10个meQTL,但绝大多数是trans-meQTL,只有一个CpG位点(SSC12:44 254 675 bp)鉴定到一个cis-meQTL,表明该位点可能受到近距离SNP调控。(4)孟德尔随机化分析显示该CpG位点(SSC12:44 254 675 bp)与b45min表型存在一定的因果关联。(5)对该位点注释发现,距离CpG位点(SSC12:44 254 675 bp)和其cis-meQTL最近的基因是NOS2,且CpG位点位于NOS2基因内。【结论】综合DNA甲基化组合基因组数据联合分析结果,可以推测NOS2基因是肉色性状关键候选基因,其DNA甲基化、SNP共同作用调控基因表达,进而影响肉色性状相关基因表达。
表观基因组关联分析;全基因组关联分析;肉质性状;DNA甲基化;甲基化数量性状位点
0 引言
【研究意义】猪的肉质性状是重要的经济性状,是肉品的营养学、卫生学、组织学和加工学特性的总和[1]。肉质性状既包含了客观特征如pH值、肉色、系水力等,也包括主观特征如风味、口感等[2]。猪肉的肉质好坏直接决定猪肉在市场的接受程度。因此,研究影响各类肉质性状的分子机制,发掘关键基因,从而指导猪的遗传改良,对改善猪肉品质具有重要意义。【前人研究进展】国内外众多研究者对影响肉质性状的基因展开大量研究,发现了一些对肉质有明显影响的主效基因,如氟烷基因[3](halothane,HAL)、酸肉基因[4](rendement napole,RN)、RKAG3[5](AMP-activated protein kinase γ 3)等。此外也鉴定了与猪肉质性状相关的重要候选基因,如瘦素基因[6-7]、脂肪酸结合蛋白基因家族[8](fatty acid binding protein,FABP)、FASN[9-10]编码脂肪酸合成酶、MYOD[11-13]基因家族等。研究者通过全基因组关联分析(genome- wide association study,GWAS)对肉质性状的遗传机制进行解析[14]。Gao等[15]对582头商业猪中对pH值、肉色、大理石花纹、导电率等肉质性状进行GWAS分析,研究发现一些关键的候选基因如与pH值相关的PPP1R3B基因、与肉色相关的SLC15A4等。Liu等[16]对杜洛克和二花脸猪F2群体进行GWAS分析,研究发现了GALNT15、GALNTL2和HTATIP2等与pH值和滴水损失显著相关的候选基因。然而,猪的肉质性状是由多基因控制的复杂性状,单一组学数据不能全面揭示影响肉质性状的分子机制。近些年来,表观遗传学和甲基化测序技术的发展为畜禽重要经济性状的表观遗传研究提供基础,例如猪[17]、牛[18]、鸡[19]、绵羊[20]和山羊[21]等。目前,对猪肉质性状的DNA甲基化研究主要集中于不同品种不同组织的全基因组DNA甲基化差异的比较分析。唐韶青等[22]发现,在同一个物种的不同组织中,其DNA甲基化模式存在特异性,其中组织的甲基化水平一般要高于血液。肌肉组织和脂肪组织是研究肉质性状最重要的组织。LI等[23]基于长白猪、藏猪和荣昌猪2个部位的肌肉组织和8个部位脂肪组织的甲基化测序数据,描绘了猪肌肉和脂肪组织的甲基化图谱,揭示了不同品种间和不同组织间的差异甲基化位点,并证实了甲基化在肥胖的发生过程中起重要作用。张小丽[24]对3头长白猪不同部位的脂肪组织进行全基因组DNA甲基化分析,研究发现基因外显子区有更高的甲基化程度,而基因启动子区域的甲基化会抑制基因的转录。总体而言,研究者对猪肉质性状的研究主要集中于基于DNA的基因组研究,而对肉质性状的DNA甲基化研究以及结合基因组和甲基化组的综合分析却鲜有报道。【本研究切入点】多项研究表明遗传变异,通常为单核苷酸多态性(single nucleotide polymorphism, SNP),可以对DNA甲基化产生重大影响[25],这些变异位点即为甲基化数量性状位点(methylation quantitative trait loci,meQTL)。通过meQTL可以探究复杂性状潜在的生物学机制,而顺式meQTL(cis-meQTL)的识别是大多数研究的热点。【拟解决的关键问题】基于纯种大白猪背最长肌的DNA甲基化组和基因组数据,利用表观基因组关联分析(epigenome-wide association analysis, EWAS)和全基因组关联分析鉴定了与肉质性状相关的关键位点。随后对GWAS鉴定的显著SNPs与EWAS鉴定的显著CpG位点进行比较分析,通过条件关联分析筛选具有独立显著信号的CpG位点,再进行meQTL鉴定和孟德尔随机化验证,最后鉴定了影响肉质的潜在关键基因。本研究为应用甲基化组和基因组联合分析,筛选肉质性状关键基因提供了新的见解。
1 材料与方法
1.1 试验动物与表型测定
选取来自浙江天蓬猪场的140头纯种大白猪(包括85头公猪和55头母猪)。所有试验猪在相同条件及环境的圈舍中饲养,并且执行相同的饲养标准。试验猪只屠宰时体重为(111.71±12.92)kg,在屠宰前禁食24 h。在屠宰时收集每头猪的左半边胴体第3—4肋处背最长肌样品,并测定pH值、肉色、滴水损失、肌内脂肪、脂肪酸组成等肉质性状。肉质性状的测定参考李娜[26]、秦一禾[27]以及张俊杰[28]的学位论文。测定具体性状包括:屠宰45 min pH值(pH45min)、屠宰24 h pH值(pH24h)、屠宰45 min亮度值(L45min)、屠宰45 min红度值(a45min)、屠宰45 min黄度值(b45min)、屠宰24 h亮度值(L24h)、屠宰24 h红度值(a24h)、屠宰24 h黄度值(b24h)、滴水损失(DL)、剪切力(SF)、肌内脂肪(IMF)、癸酸(C10:0)、月桂酸(C12:0)、肉蔻酸(C14:0)、棕榈酸(C16:0)、硬脂酸(C18:0)、花生酸(C20:0)、棕榈油酸(C16:1)、油酸(C18:1n-9)、顺式-11-二十碳一烯酸(C20:1n-9)、亚油酸(C18:2n-6)、α-亚麻酸(C18:3n-3)、γ-亚麻酸(C18:3n-6)、顺式-11,14-二十碳二烯酸(C20:2)、顺式-11,14,17-二十碳三烯酸(C20:3n-3)、顺式-8,11,14-二十碳三烯酸(C20:3n-6)、花生四烯酸(C20:4n-6)、二十二碳六烯酸(C22:6n-3)。
1.2 组织样本采集及DNA提取
在对140头试验猪进行屠宰测定的同时采集背最长肌样品于冻存管中,加入75%的乙醇,于-20 ℃条件下保存备用。样品DNA的提取参照OMEGA组织DNA提取试剂盒说明书所述步骤进行。
1.3 简化甲基化测序及质量控制
简化甲基化测序(reduced representation bisulfite sequencing,RRBS)是一种准确且经济的DNA甲基化测序方案。RRBS通过酶切MspI(methylation- insensitive restriction enzyme)富集启动子及CpG岛区域,并进行Bisulfite测序。本研究采集140头大白猪的背最长肌样品,基于RRBS技术进行甲基化测序。首先检测DNA样品质量,通过琼脂糖凝胶电泳结果判断DNA降解程度以及是否有RNA污染,然后利用Nanodrop检测DNA浓度,最后利用Qubit对DNA浓度精确定量。合格的DNA样品加入一定比例的阴性对照(lambda DNA),接着使用MspI对DNA样品进行酶切处理(37 ℃,16 h)。随后对酶切后的DNA片段采用末端修复、加A尾,并连上胞嘧啶均经过甲基化修饰的全部测序接头。通过切胶选择插入片段长度在40—220 bp范围的DNA片段进行Bisulfite处理,经处理后没有发生甲基化的C变为U(经过PCR扩增后变为T),而甲基化的C保持不变。最后进行PCR扩增,获得最终的DNA文库。文库构建完成后进行质量检验,使用Qubit2.0对构建的文库进行初步定量,将文库稀释至1 ng·µL-1,随后使用Agilent 2100检测文库的插入片段长度,达到预期片段长度后,使用qPCR方法对文库的有效浓度进行准确定量,保证文库质量,文库质量检验合格后,进行上机测序。甲基化测序交由北京诺禾致源科技公司完成。
测序完成获得原始下机数据raw data,对raw data进行初步过滤以去除质量差的数据,使用Trimmomatic软件[29]对raw data过滤,去除测序数据的测序接头和低质量片段,从而获得高质量clean data。首先采用滑动窗口的方式去除低质量reads,4个碱基为一个窗口,若该窗口平均碱基质量低于15,则在此截去reads,参数设置为SLIDINGWINDOW: 4:15。去除尾部质量小于3或者包含无法确定的碱基信息的reads,参数设置为TRAILING:3。随后去除接头污染的reads,去除接头有两种模式,包括simple alignment mode(seed与接头序列比对得分超过7)的模式和palindrome mode(当read1与read2的重叠区域碱基评分超过30时,截去seed部分序列)的模式,参数设置为ILLUMINACLIP:adapter.fa:2:30:7:1:true。最后去除不能形成paired的reads。
经过初步过滤后获得clean data被用于后续分析,然后使用FastQC软件对clean data进行再次质量统计,以保证数据质量。数据质量合格后,将clean data比对到猪参考基因组上(sus scrofa 11.1)上进行分析。使用Bismark v0.22.1软件[30]的bismark_genome_ preparation函数准备参考基因组;调用bowtie2 v2.3.5.1软件[31]对双端测序文件进行比对,生成BAM文件;使用Bismark v0.22.1软件的deduplicate bismark函数对BAM文件去除重复序列;使用Bismark v0.22.1软件的bismark methylation extractor函数提取甲基化统计信息文件。提取的甲基化统计信息文件中包含覆盖率信息以及在一个基因组位置上甲基化reads和未甲基化reads的比例信息。
1.4 表观基因组关联分析
在EWAS分析之前,首先对每个个体的甲基化统计信息文件进行质量控制以获得用于EWAS分析的高质量位点。质量控制标准包括:在所有个体中只保留CpG位点;每个个体中只保留覆盖度大于10 reads的位点;所有个体中保留至少在30个个体中检出的CpG位点。过滤后,基于R软件包CpGassoc v2.60对CpG位点甲基化水平和肉质性状进行关联分析。在EWAS分析时,首先使用Box-Cox转换将表型数据校正至符合正态分布,然后用出生年、出生月、性别和批次作为固定因子对表型进行校正,获得表型校正值,然后应用混合线性模型进行关联分析,模型如下:
=+++
模型中,表示表型向量;表示平均值;表示CpG甲基化水平矩阵;表示CpG效应;表示随机效应;表示残差,服从(~(0,2))分布,表示单位向量。
使用Bonferroni校正法来设置全基因组显著阈值,即EWAS结果中当CpG位点的关联值达到<0.05/时,表示该位点为显著位点,其中表示EWAS分析中使用的CpG位点数量。
1.5 SNP分型和基因型填充
基于Illumina平台(Illumina,美国)标准流程使用Neogen GeneSeek Porcine50K芯片对140头大白猪进行基因分型,芯片包含50 697个SNPs,随后对芯片数据进行填充。基因型填充参考模板是本课题获取的40头大白猪重测序数据。对重测序的原始数据使用FastQC软件进行严格的质量控制以获得clean data,然后对clean data进行序列比对和SNP检测。使用BWA v0.7.17软件[32]将clean reads比对到猪参考基因组上(sus scrofa 11.1),设置比对参数mem -t 4 -k 32 -M;使用SAMtools v1.19软件[33]将比对获得的SAM格式文件转换为BAM格式文件并进行排序;使用Picard v1.119软件的MarkerDuplicates去除BAM文件中的PCR重复序列;使用GATK v4.1.9.0软件[34]的HaplotypeCaller函数对BAM文件进行变异检测,从而获得原始SNP信息;使用GATK v4.1.9.0软件对原始SNP信息文件进行质量控制以去除结果中的假阳性位点,过滤标准为:QualByDepth (QD)<2.0,FisherStrand(FS)<60.0,RMSMappingQuality(MQ)<40.0,MappingQualityRankSumTest<-12.5以及ReadPosRankSumTest<-8.0。
填充完成后利用 Plink v1.09软件[35]对填充后的SNP数据进行质量控制以获得高质量SNP位点,过滤标准为:去除实际等位基因与最可能等位基因相关性平方(R2)小于0.9的SNP位点;去除MAF小于0.01的SNP位点;去除HWE检验小于1×10-6的SNP位点;去除性染色体上的SNP位点。经过质量控制的SNP位点被用于后续GWAS分析。
1.6 全基因组关联分析
使用GEMMA v0.98.5软件[36]中的线性混合模型对28个肉质性状进行GWAS分析。在GWAS分析前,先使用Box-Cox变换将表型数据校正至符合正态分布,然后以出生年、出生月、性别、批次作为固定因子对表型进行校正,得到的校正值被用于GWAS分析。GWAS所使用的线性混合模型如下:
=++
模型中,表示表型值向量;表示SNP效应;表示剩余多基因效应,服从(~(0,2))分布,表示分子亲缘关系矩阵(genomic relatedness matrix,GRM);表示残差,服从(~(0,2))分布,表示单位向量;和分别表示和的关联矩阵。GRM根据如下公式计算:
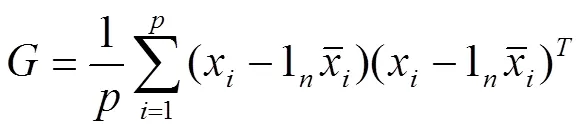
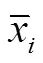
使用Bonferroni校正法来设置全基因组显著阈值,即GWAS结果中当SNP的关联值达到<0.05/时,表示该SNP为显著SNP位点,其中表示GWAS分析中使用的SNP数量;建议显著阈值设置为<1/。使用R软件中GenABEL包[37]中estlambda函数计算基因组膨胀系数λ用于判断是否有假阳性信号。
1.7 鉴定EWAS与GWAS重叠的显著关联位点
本研究首先对140头试验大白猪进行50K芯片基因分型,经质量控制和基因型填充后进行GWAS分析。同时基于RRBS对所有个体进行了甲基化测序,经过质量控制之后用于EWAS分析。相关结果发表在意大利动物科学杂志[38]。本文对肉质性状的GWAS鉴定的显著SNPs与EWAS鉴定的显著CpG位点进行比较,从而鉴定GWAS与EWAS重叠的显著关联位点。如果GWAS的显著SNPs与EWAS显著CpG位点在2 Mb范围内,则被认为是二者重叠的显著关联位点[39]。
1.8 条件关联分析
为了确定GWAS与EWAS重叠的显著关联位点是由SNPs和CpG共同导致或者是由同一位置的SNPs或CpG的独立信号导致,基于GWAS与EWAS重叠的显著关联,通过条件关联分析,使用重叠的显著关联位点的SNP作为协变量重新进行EWAS分析[39]。模型如下:
=++++
模型中,表示表型向量;表示平均值;表示CpG甲基化水平矩阵;表示CpG效应;表示SNP取值;表示SNP效应;表示随机效应;表示残差,服从(~(0,2))分布,表示单位向量。
使用Bonferroni校正法来设置全基因组显著阈值,即EWAS结果中当CpG位点的关联值达到<0.05/时,表示该位点为显著位点,其中表示EWAS分析中使用的CpG位点数量。如果使用SNP作为协变量重新进行EWAS分析后,原本显著的CpG位点的显著性消失,表明该EWAS中CpG位点的显著性受到GWAS结果的混淆;如果重新进行EWAS分析后,该CpG位点仍保持显著性,表明该重叠的显著关联位点是由CpG位点的显著信号产生的,EWAS鉴定的显著CpG位点没有受到GWAS的混淆。
1.9 鉴定甲基化数量性状位点
以SNP为自变量,以条件关联分析中仍保持显著的CpG位点的甲基化水平作为因变量并进行关联分析,从而鉴定甲基化数量性状位点(meQTL)。MeQTL可以根据遗传变异与CpG位点的距离而分为顺式meQTL(cis-meQTL)和反式meQTL(trans-meQTL)。其中,cis-meQTL是靠近CpG位点的遗传变异(二者距离在1 Mb范围内),而trans-meQTL是与CpG位点距离较远(二者距离超过1 Mb)或者位于不同染色体的遗传变异[40]。在关联分析中使用混合线性模型,具体模型如下:
=++
模型中,表示CpG甲基化水平;表示基因型向量;表示SNP效应;表示剩余多基因效应的关系矩阵;表示剩余多基因效应;表示残差,服从(~(0,2))分布,表示单位向量。
使用Bonferroni校正法来设置全基因组显著阈值,即关联结果中当SNP的值达到<0.05/时,表示该SNP为显著SNP位点,其中表示关联分析中使用的SNP数量,即全基因组显著阈值为<0.05/10961097。
1.10 孟德尔随机化分析
孟德尔随机化是指个体的遗传变异导致不同程度的DNA甲基化或不同水平的特定表型[41]。使用单样本孟德尔随机化分析来将-meQTL与EWAS整合分析,可以推断-meQTL的位点和表型之间是否存在因果关系。使用R软件的MendelianRandomization v0.5.0包中的逆加权方差方法(INVERSE-variance weighted,IVW)对-meQTL的位点进行孟德尔随机化分析。其中SNP与表型性状关联的效应值(β)和标准误(se(β))以及SNP与CpG位点关联的效应值(β)和标准误(se(β))由关联分析结果获得。
2 结果
2.1 GWAS和EWAS重叠的显著关联位点
为了从基因组水平验证EWAS结果,首先对EWAS显著CpG位点和GWAS显著SNP位点进行比较分析。如果GWAS的显著SNP位点与EWAS显著CpG位点距离在2 Mb范围内,则被认为是GWAS与EWAS重叠的显著关联位点[38]。基于肉质性状的EWAS和GWAS结果,在28个肉质性状中,共在3个性状(b45min、DL和C22:6n-3)上发现了GWAS与EWAS重叠的显著关联位点(表1)。其中,在b45min中发现了8个显著CpG位点和59个显著SNP位点位置接近;在DL发现1个CpG位点和1个SNP位点位置接近;在C22:6n-3中发现了3个CpG位点与146个SNP位点位置接近。这些位点被确定为重叠的显著关联位点。

表1 GWAS和EWAS重叠的显著关联信号统计
2.2 条件关联分析
为了确定GWAS与EWAS重叠的显著关联位点是由SNPs和CpG共同导致或者是由同一位置的SNPs或CpG的独立信号导致,使用 SNP 作为协变量构建混合线性模型对其中的 CpG 重新进行条件关联分析。条件关联分析结果显示:对于 b45min性状,在8个显著的CpG位点中,有7个CpG位点在经过条件关联分析后仍保持显著(<6.25×10−4),表明这7个CpG位点的显著不受附近的 GWAS 显著SNP 的影响;对于DL性状,1个CpG位点在经过条件关联分析后仍达到显著阈值(<0.05),表明该位点的显著关联不受附近的显著SNP影响;对于C22:6n-3性状,3个CpG位点在经过条件关联分析后未能达到显著阈值(>1.67×10−2),表明这些CpG位点的显著可能是受到GWAS显著SNP影响导致的假阳性(表2)。
2.3 鉴定甲基化数量性状位点
随后使用条件关联分析中仍保持显著的CpG位点的甲基化水平作为因变量,以SNP作为自变量进行全基因组关联分析鉴定meQTL,即使用b45min性状的7个CpG位点(SSC1:4 667 485 bp、SSC7:10 489 621 bp、SSC10:36 329 538 bp、SSC12:44 254 675 bp、SSC14: 17 859 513 bp、SSC18:48 429 425 bp和SSC18:50 979 305 bp)和DL性状的1个CpG位点(SSC7:120 954 182 bp)的甲基化水平作为因变量进行关联分析从而鉴定meQTL(表3)。
在b45min性状的7个CpG位点中,在其中3个位点中(SSC1:4 667 485 bp位点(图1-A)、SSC10: 36 329 538 bp(图1-C)和SSC18:48 429 425 bp(图1-F))没有识别到显著关联。对于SSC7:10 489 621 bp(图1-B)位点,鉴定了2个meQTL,均为trans-meQTL,对应9个显著SNP位点。对于SSC12:44 254 675 bp(图1-D)位点,鉴定了2个meQTL,包括1个cis-meQTL和1个trans-meQTL,对应11个显著SNP位点。对于SSC14:17 859 513 bp(图1-E)位点,鉴定了2个meQTL,2个均为trans-meQTL,对应46个显著SNP位点。对于SSC18:50 979 305 bp(图1-G)位点,GWAS鉴定了3个meQTL, 3个均为trans-meQTL,对应345个显著SNP位点。对于DL性状的SSC7: 120 954 182 bp(图1-H)位点中,鉴定了1个trans- meQTL ,对应4个显著SNP位点。在鉴定的meQTL中,只有一个cis-meQTL,其SNP位点(SSC12: 43 262 418 bp)位于CpG位点(SSC12:44 254 675 bp)上游约1 Mb的区域,表明该位点可能受到近距离 SNP 调控。该cis-meQTL的SNP基因型与CpG位点甲基化水平如图2-A。

表2 对GWAS与EWAS共同的显著信号条件关联分析结果
value (after correct)是指条件关联分析的值
value (after correct) refers to thevalue of conditional association analysis
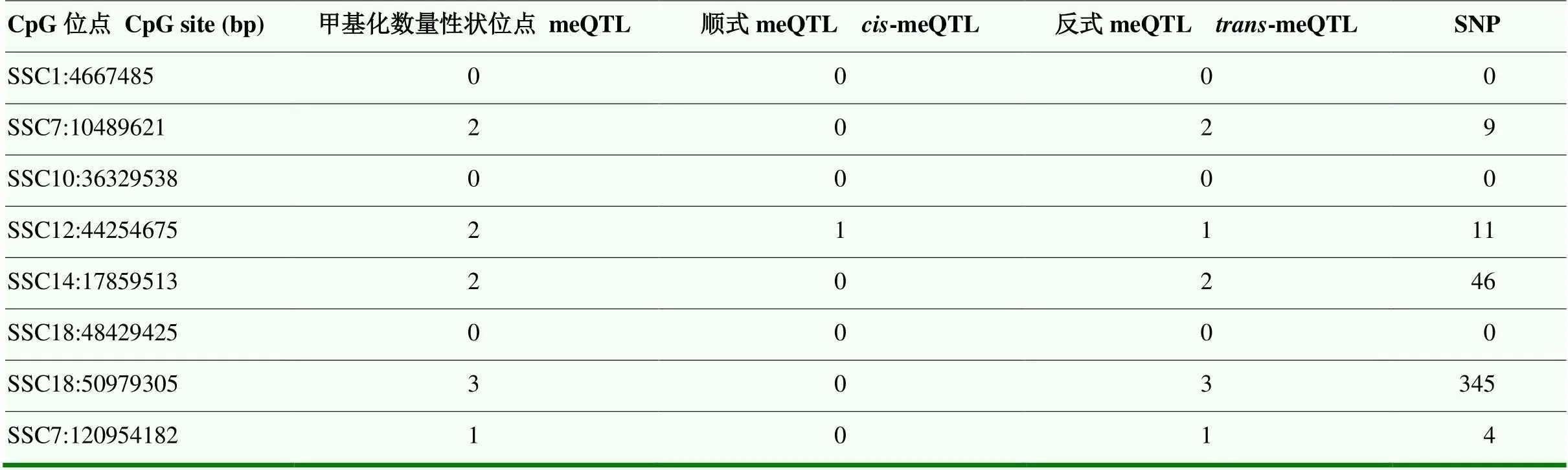
表3 甲基化数量性状位点鉴定结果
2.4 孟德尔随机化分析
为了进一步估计DNA甲基化在SNP和表型性状关联的中介作用,我们对鉴定的cis-meQTL的SNP位点(SSC12:43 262 418 bp)和对应CpG位点(SSC12:44 254 675 bp)进行孟德尔随机化分析。使用cis-meQTL作为工具变量,对应CpG位点(SSC12:44 254 675 bp)作为介质变量,表型性状(b45min)作为结果变量进行孟德尔随机化分析,发现SNP对CpG位点甲基化的影响与SNP对表型性状的影响存在一个线性关系(图2-B),表明该CpG位点与屠宰45 min黄度值(b45min)存在一定的因果关联。
随后对该位点注释发现,距离 CpG 位点(SSC12: 44 254 675 bp)和其cis-meQTL 最近的基因是 NOS2,且CpG位点位于 NOS2 基因内。综合DNA甲基化组和基因组数据联合分析结果,本研究推测NOS2 基因可能是肉色性状关键候选基因,其 DNA 甲基化、SNP 共同作用调控基因表达,进而影响肉色性状相关基因表达。
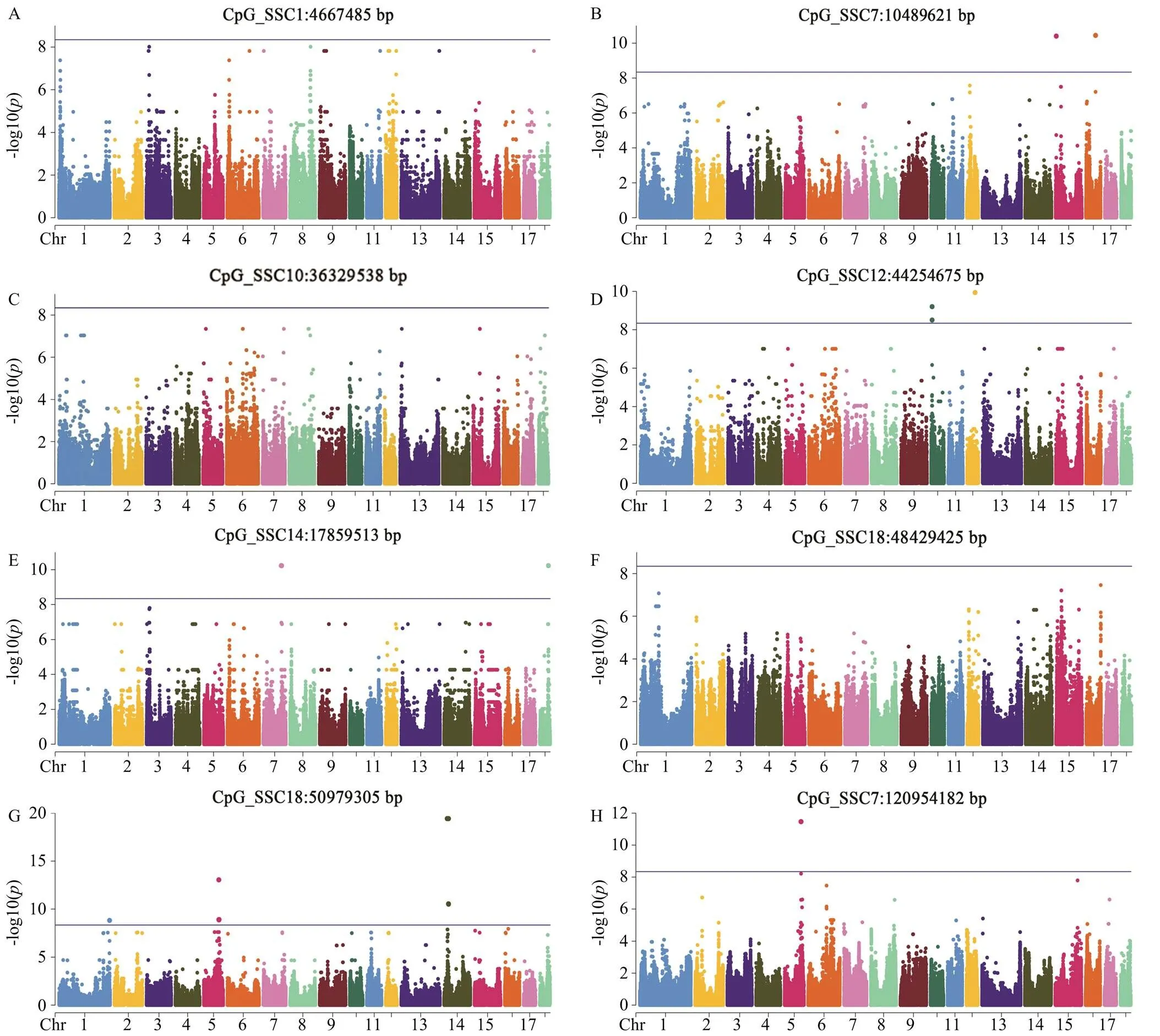
图1 CpG位点与基因组SNP关联分析的曼哈顿图
3 讨论
本研究通过整合DNA甲基化组和全基因组数据对肉质性状进行EWAS分析和GWAS分析。EWAS鉴定了肉质性状与DNA甲基化之间的3 107个显著关联,GWAS鉴定了肉质性状与SNP之间的514个显著关联。将EWAS结果与GWAS结果联合分析发现,在屠宰45 min黄度值(b45min)、滴水损失(DL)和二十二碳六烯酸(C22:6n-3)上EWAS和GWAS都在同一基因组区域鉴定到显著关联,即EWAS鉴定的显著CpG位点和GWAS鉴定的显著SNP位点在基因组的位置彼此接近。因此本研究通过筛选GWAS的显著SNP位点与EWAS显著CpG位点距离在2 Mb范围内重叠的显著关联位点进行进一步分析。经过重叠信号的比较分析后,在b45min中发现了8个显著CpG位点和59个显著SNP位点位置接近;在DL发现1个CpG位点和1个SNP位点位置接近;在C22:6n-3中发现了3个CpG位点与146个SNP位点位置接近。这些位点被确定为重叠的显著关联位点,也是本研究进行下一步甲基化数量性状位点鉴定的基础。
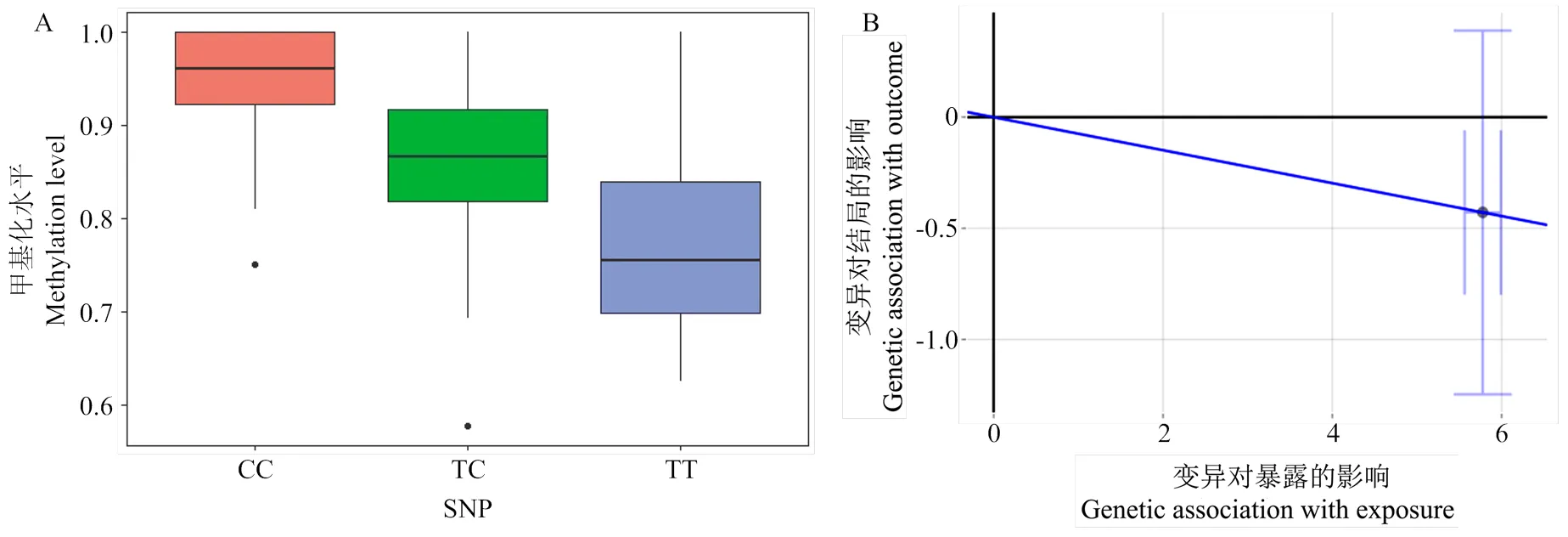
A. CpG位点SSC12:44 254 675 bp的cis-meQTL的箱线图,横坐标表示SNP基因型,纵坐标表示CpG甲基化水平;B. cis-meQTL的孟德尔随机化分析的散点图,横坐标表示SNP位点(SSC12:43 262 418 bp)对CpG 位点(SSC12:44 254 675 bp)甲基化的影响,纵坐标表示SNP位点(SSC12: 43 262 418 bp)对表型性状(b45min)的影响
3.1 条件关联分析
基因表达受多种调控方式影响,DNA甲基化和SNP都可以影响基因的表达,有许多研究报道了SNP、甲基化和基因表达的关系[25-42]。因此,一些EWAS的显著信号可能是由附近的SNP导致的,由SNP或者其他因素影响引起的这种混淆可能是EWAS鉴定到大量与性状相关的显著DNA甲基化的主要驱动因素。这一结果也在许多人类研究中被报道。例如,SHEN等[38]通过对人类疾病蛋白质生物标志物进行GWAS和EWAS并综合比较分析后,发现与GWAS重叠的EWAS显著信号是由潜在的遗传变异以及环境因素引起的。因此,本文通过以SNP为协变量对这些EWAS与GWAS重叠的显著关联位点进行条件关联分析发现,在b45min、DL和C22:6n-3这3个性状中,b45min的7个CpG位点在条件关联分析后仍保持显著,DL的1个CpG位点在条件关联分析后仍保持显著,表明这些CpG位点关联的显著性是独立的,不受GWAS显著SNP的影响;而C22:6n-3的3个位点在使用SNP作为协变量分析后不再显著,表明这些CpG位点的关联显著性可能受到附近SNP的影响而产生的假阳性。
3.2 孟德尔随机化分析
EWAS旨在系统地将整个表观基因组中的DNA甲基化水平的变异与表型变异关联起来。多项研究显示遗传变异(通常为SNPs)可以对DNA甲基化产生重大影响[25],这些位点即为甲基化数量性状位点(methylation quantitative trait loci,MeQTL)。MeQTL可以根据遗传变异与CpG位点的距离而分为顺式meQTL(cis-meQTL)和反式meQTL(trans- meQTL)。本文对b45min的7个CpG位点以及DL的1个CpG位点,以CpG位点的甲基化水平为因变量,以SNP为自变量进行关联分析,从而鉴定meQTL。结果发现了10个meQTL,但绝大多数是trans-meQTL,只有一个CpG位点(SSC12:44 254 675 bp)鉴定到一个cis-meQTL,表明该位点可能受到近距离SNP调控。由于meQTL潜在的混杂作用,可能会造成DNA甲基化水平与表型之间出现显著关联,并且大多数研究中并没有考虑meQTL。因此,有研究者提出在进行EWAS之前利用meQTL效应来校正DNA甲基化值以解决这个问题[43-44],从而将meQTL整合到EWAS中。在EWAS中整合meQTL的另一种应用是通过孟德尔随机化深入分析DNA甲基化信号与相关表型之间关联的假定因果方向[45]。在表观遗传分析的背景下,将meQTL作为工具变量,DNA甲基化水平作为暴露因素,而表型性状作为结果[46]。已经有多项研究应用孟德尔随机化将meQTL整合到EWAS分析中[39,47-48]。此外,EWAS和meQTL信号的组合也可以用于探索遗传-环境的相互作用[49]。因此,通过EWAS鉴定的与性状相关的CpG位点有可能反映性状对DNA甲基化的影响,而不是DNA甲基化对性状的作用[48-50]。基于此,本文使用cis-meQTL作为工具变量进行孟德尔随机化分析,结果表明了DNA甲基化对性状的影响。
3.3 NOS2基因
距离CpG位点(SSC12:44 254 675 bp)和其cis- meQTL最近的基因是NOS2,且CpG位点位于NOS2基因内。在该基因组区域,Choi等[51]也鉴定到与肉色b值相关的QTL区域,此外,Kim等[52]也在该区域鉴定到与肉色L值相关的QTL区域。猪肌肉色素主要由肌红蛋白、血红蛋白以及微量有色代谢物组成。在放血充分的情况下,对肉色起主要作用的是肌红蛋白。NOS2基因属于一氧化氮合酶家族,一氧化氮合酶催化L-精氨酸产生NO和L-瓜氨酸[53]。Hattori等[54]研究表明在脂多糖和细胞因子刺激的血管平滑肌细胞中,PI3K-Akt通路可以通过NF-kappa B(核因子kappa B)调控iNOS的表达。Silva等[55]在小鼠上的研究发现ATP通过刺激PI3激酶和Akt来增强一氧化氮酶的活性,进而增加NO的产生。NO是一种信使分子,在许多组织、细胞和器官中的核心生物过程中作为关键的调节分子。NO也可以与OxyMb反应形成MetMb和硝酸盐[56],NO对线粒体呼吸也有影响,NO可以抑制线粒体呼吸链[57],导致ATP产生减少,氧化剂产生增加,从而减少线粒体对氧气的消耗。有研究表明,肌红蛋白也可以通过氧化、配体结合以及形成生物活性S-亚硝基肌红蛋白的方式调节NO平衡[58]。因此NOS2基因可能通过影响细胞内NO的浓度进而影响肉色性状。
4 结论
利用140头大白猪背最长肌的28个肉质性状EWAS分析和GWAS分析的结果,筛选了GWAS的显著SNP位点与EWAS显著CpG位点重叠的显著关联位点,通过条件关联分析筛选了具有独立效应的显著位点,随后进行了meQTL鉴定和孟德尔随机化分析。结果表明CpG位点(SSC12:44 254 675 bp)鉴定的cis-meQTL与屠宰45 min黄度值(b45min)存在一定的因果关联,且该位点位于NOS2基因内。因此,本论文推测NOS2基因可能是肉色性状关键候选基因,其DNA甲基化、SNP共同作用调控基因表达,进而影响肉色性状相关基因表达,但其在肉色性状调控的具体分子机制需进一步试验研究。
[1] 孙建广, 张石蕊. 猪肉品质研究进展. 中国猪业, 2008, 3(7): 58-59.
SUN J G, ZHANG S R. Research progress of pork quality. China Swine Industry, 2008, 3(7): 58-59. (in Chinese)
[2] 宋志芳, 解幼志, 刑荷岩, 芦春莲, 曹洪战. 影响猪肉质性状相关基因的研究进展. 中国猪业, 2017, 12(7): 48-50.
SONG Z F, XIE Y Z, XING H Y, LU C L, CAO H Z. Research progress on genes related to pork quality traits. China Swine Industry, 2017, 12(7): 48-50. (in Chinese)
[3] SHEN Q W, UNDERWOOD K R, MEANS W J, MCCORMICK R J, DU M. The halothane gene, energy metabolism, adenosine monophosphate-activated protein kinase, and glycolysis in postmortem pig longissimus dorsi muscle. Journal of Animal Science, 2007, 85(4): 1054-1061.
[4] SALAS R C D, MINGALA C N. Genetic factors affecting pork quality: halothane and rendement napole genes. Animal Biotechnology, 2017, 28(2): 148-155.
[5] 李梦云, 郑萍, 李婉涛, 陈代文. PRKAG3基因在不同品种猪不同生长阶段骨骼肌中的表达差异及其表达量与肉质的关系. 动物营养学报, 2017, 29(5): 1661-1669.
LI M Y, ZHENG P, LI W T, CHEN D W. The expression difference ofgene in skeletal muscle at different growth stages for different breeds of pigs and the relationship betweengene expression level and meat quality. Chinese Journal of Animal Nutrition, 2017, 29(5): 1661-1669. (in Chinese)
[6] MÜNZBERG H. Differential leptin access into the brain-a hierarchical organization of hypothalamic leptin target sites? Physiology & Behavior, 2008, 94(5): 664-669.
[7] 于太永. 脂肪细胞因子leptin和TNF: α对猪骨骼肌成肌细胞增殖分化的影响及其机制[D]. 杨凌: 西北农林科技大学, 2007.
YU T Y. Effects of adipocytokines leptin and TNF-α on the proliferation and differentiation of porcine skeletal myoblasts and its mechanism[D]. Yangling: Northwest A & F University, 2007. (in Chinese)
[8] 李志娟. 猪心脏型脂肪酸结合蛋白研究进展. 养猪, 2021(2): 55-56.
LI Z J. Research progress of heart fatty acid-binding protein. Swine Production, 2021(2): 55-56. (in Chinese)
[9] GRZES M, SADKOWSKI S, RZEWUSKA K, SZYDLOWSKI M, SWITONSKI M. Pig fatness in relation to FASN andgenes polymorphism and their transcript level. Molecular Biology Reports, 2016, 43(5): 381-389.
[10] ZAPPATERRA M, LUISE D, ZAMBONELLI P, MELE M, SERRA A, COSTA L N, DAVOLI R. Association study between backfat fatty acid composition and SNPs in candidate genes highlights the effect ofpolymorphism in large white pigs. Meat Science, 2019, 156: 75-84.
[11] VERNER J, HUMPOLÍČEK P, KNOLL A. Impact of MYOD family genes on pork traits in Large White and Landrace pigs. Journal of Animal Breeding and Genetics, 2007, 124(2): 81-85.
[12] KIM J M, CHOI B D, KIM B C, PARK S S, HONG K C. Associations of the variation in the porcine myogenin gene with muscle fibre characteristics, lean meat production and meat quality traits. Journal of Animal Breeding and Genetics = Zeitschrift Fur Tierzuchtung Und Zuchtungsbiologie, 2009, 126(2): 134-141.
[13] LEE E A, KIM J M, LIM K S, RYU Y C, JEON W M, HONG K C. Effects of variation in porcinegene on muscle fiber characteristics, lean meat production, and meat quality traits. Meat Science, 2012, 92(1): 36-43.
[14] 周李生, 杨杰, 刘先先, 张志燕, 杨斌, 麻骏武. 苏太猪宰后72 h pH和肉色性状的全基因组关联分析. 中国农业科学, 2014, 47(3): 564-573. doi: 10.3864/j.issn.0578-1752.2014.03.016.
ZHOU L S, YANG J, LIU X X, ZHANG Z Y, YANG B, MA J W. Genome-wide association analyses for musle pH 72 h value and meat color traits in sutai pigs. Scientia Agricultura Sinica, 2014, 47(3): 564-573. doi: 10.3864/j.issn.0578-1752.2014.03.016.(in Chinese)
[15] GAO G X, GAO N, LI S C, KUANG W J, ZHU L, JIANG W, YU W W, GUO J B, LI Z L, YANG C Z, ZHAO Y X. Genome-wide association study of meat quality traits in a three-way crossbred commercial pig population. Frontiers in Genetics, 2021, 12: 614087.
[16] LIU X X, ZHANG J J, XIONG X W, CHEN C Y, XING Y Y, DUAN Y Y, XIAO S J, YANG B, MA J W. An integrative analysis of transcriptome and GWAS data to identify potential candidate genes influencing meat quality traits in pigs. Frontiers in Genetics, 2021, 12: 748070.
[17] YANG Y L, FAN X H, YAN J Y, CHEN M Y, ZHU M, TANG Y J, LIU S Y, TANG Z L. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Research, 2021, 49(3): 1313-1329.
[18] HALUŠKOVÁ J, HOLEČKOVÁ B, STANIČOVÁ J. DNA methylation studies in cattle. Journal of Applied Genetics, 2021, 62(1): 121-136.
[19] RAN J S, LI J J, YIN L Q, ZHANG D H, YU C L, DU H R, JIANG X S, YANG C W, LIU Y P. Comparative analysis of skeletal muscle DNA methylation and transcriptome of the chicken embryo at different developmental stages. Frontiers in Physiology, 2021, 12: 697121.
[20] FAN Y X, LIANG Y X, DENG K P, ZHANG Z, ZHANG G M, ZHANG Y L, WANG F. Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genomics, 2020, 21(1): 327.
[21] KANG B N, WANG J J, ZHANG H F, SHEN W, EL-MAHDY OTHMAN O, ZHAO Y, MIN L J. Genome-wide profile in DNA methylation in goat ovaries of two different litter size populations. Journal of Animal Physiology and Animal Nutrition, 2022, 106(2): 239-249.
[22] 唐韶青, 张沅, 徐青, 孙东晓, 俞英. 不同动物部分组织基因组甲基化程度的差异分析. 农业生物技术学报, 2006, 14(4): 507-510.
TANG S Q, ZHANG Y, XU Q, SUN D X, YU Y. Analysis of methylation level of genome in various tissues of different animal species. Journal of Agricultural Biotechnology, 2006, 14(4): 507-510. (in Chinese)
[23] LI M Z, WU H L, LUO Z G, XIA Y D, GUAN J Q, WANG T, GU Y R, CHEN L, ZHANG K, MA J D, et al. An atlas of DNA methylomes in porcine adipose and muscle tissues. Nature Communications, 2012, 3: 850.
[24] 张小丽. 猪背部浅层和背部深层脂肪组织全基因组甲基化研究[D]. 雅安: 四川农业大学, 2013.
ZHANG X L. Genome-wide DNA methylation changes between the superficial and deep backfat tissues of the pig[D]. Yaan: Sichuan Agricultural University, 2013. (in Chinese)
[25] VILLICAÑA S, BELL J T. Genetic impacts on DNA methylation: research findings and future perspectives. Genome Biology, 2021, 22(1): 127.
[26] 李娜. 基于GWAS的猪肉品质性状候选基因研究[D]. 北京: 中国农业大学, 2016.
LI N. Genome-wide association studies for pig meat traits and exploration of major genes[D]. Beijing: China Agricultural University, 2016. (in Chinese)
[27] 秦一禾. 猪肌内脂肪含量的全基因组关联研究[D]. 昆明: 云南大学, 2017.
QIN Y H. Genome-wide association studies for intramuscular fat content in swine[D]. Kunming: Yunnan University, 2017. (in Chinese)
[28] 张俊杰. 利用多品种大样本群体精细定位影响猪背最长肌脂肪酸组成的QTL[D]. 南昌: 江西农业大学, 2018.
ZHANG J J. Mapping and fine-mapping QTL affecting longissimus muscle fatty acid composition in large scale multiple pig populations[D]. Nanchang: Jiangxi Agricultural University, 2018. (in Chinese)
[29] BOLGER A M, LOHSE M, USADEL B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014, 30(15): 2114-2120.
[30] KRUEGER F, ANDREWS S R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 2011, 27(11): 1571-1572.
[31] LANGMEAD B, SALZBERG S L. Fast gapped-read alignment with Bowtie 2. Nature Methods, 2012, 9: 357-359.
[32] LI H, DURBIN R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 2009, 25(14): 1754-1760.
[33] LI H, HANDSAKER B, WYSOKER A, FENNELL T, RUAN J, HOMER N, MARTH G, ABECASIS G, DURBIN R. 1 0 0 0 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics, 2009, 25(16): 2078-2079.
[34] DEPRISTO M A, BANKS E, POPLIN R, GARIMELLA K V, MAGUIRE J R, HARTL C, PHILIPPAKIS A A, DEL ANGEL G, RIVAS M A, HANNA M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 2011, 43: 491-498.
[35] CHANG C C, CHOW C C, TELLIER L C, VATTIKUTI S, PURCELL S M, LEE J J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience, 2015, 4: 7.
[36] ZHOU X, STEPHENS M. Genome-wide efficient mixed-model analysis for association studies. Nature Genetics, 2012, 44: 821-824.
[37] AULCHENKO Y S, RIPKE S, ISAACS A, VAN DUIJN C M. GenABEL: An R library for genome-wide association analysis. Bioinformatics, 2007, 23(10): 1294-1296.
[38] SHEN Q, WANG K, WANG S J, ZHAO Z J, JI X, CHEN D, YU Y, CUI S D, WANG J G, CHEN Z Y, GU Y R, TANG G Q. Identifying key DNA methylation sites and their cis-methylation quantitative loci for intramuscular fatty acid traits using genome and methylome data in Yorkshire pigs. Italian Journal of Animal Science, 2023, 22(1): 829-840.
[39] AHSAN M, EK W E, RASK-ANDERSEN M, KARLSSON T, LIND-THOMSEN A, ENROTH S, GYLLENSTEN U, JOHANSSON Å. The relative contribution of DNA methylation and genetic variants on protein biomarkers for human diseases. PLoS Genetics, 2017, 13(9): e1007005.
[40] YU F T, QIU C, XU C, TIAN Q, ZHAO L J, WU L, DENG H W, SHEN H. Mendelian randomization identifies CpG methylation sites with mediation effects for genetic influences on BMD in peripheral blood monocytes. Frontiers in Genetics, 2020, 11: 60.
[41] BURGESS S, THOMPSON S G. Use of allele scores as instrumental variables for Mendelian randomization. International Journal of Epidemiology, 2013, 42(4): 1134-1144.
[42] ZHI D G, ASLIBEKYAN S, IRVIN M R, CLAAS S A, BORECKI I B, ORDOVAS J M, ABSHER D M, ARNETT D K. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics, 2013, 8(8): 802-806.
[43] BIRNEY E, SMITH G D, GREALLY J M. Epigenome-wide association studies and the interpretation of disease-omics. PLoS Genetics, 2016, 12(6): e1006105.
[44] LAPPALAINEN T, GREALLY J M. Associating cellular epigenetic models with human phenotypes. Nature Reviews Genetics, 2017, 18: 441-451.
[45] DAVEY SMITH G, HEMANI G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Molecular Genetics, 2014, 23(R1): R89-R98.
[46] RELTON C L, DAVEY SMITH G. Mendelian randomization: applications and limitations in epigenetic studies. Epigenomics, 2015, 7(8): 1239-1243.
[47] BATTRAM T, RICHMOND R C, BAGLIETTO L, HAYCOCK P C, PERDUCA V, BOJESEN S E, GAUNT T R, HEMANI G, GUIDA F, CARRERAS-TORRES R, et al. Appraising the causal relevance of DNA methylation for risk of lung cancer. International Journal of Epidemiology, 2019, 48(5): 1493-1504.
[48] WAHL S, DRONG A, LEHNE B, LOH M, SCOTT W R, KUNZE S, TSAI P C, RIED J S, ZHANG W H, YANG Y W, et al. Epigenome- wide association study of body mass index, and the adverse outcomes of adiposity. Nature, 2017, 541: 81-86.
[49] TSAPROUNI L G, YANG T P, BELL J, DICK K J, KANONI S, NISBET J, VIÑUELA A, GRUNDBERG E, NELSON C P, MEDURI E, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics, 2014, 9(10): 1382-1396.
[50] ZWAMBORN R A J, SLIEKER R C, MULDER P C, ZOETEMELK I, VERSCHUREN L, SUCHIMAN H E D, TOET K H, DROOG S, SLAGBOOM P E, KOOISTRA T, et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Scientific Reports, 2017, 7: 43261.
[51] CHOI I, STEIBEL J P, BATES R O, RANEY N E, RUMPH J M, ERNST C W. Identification of carcass and meat quality QTL in an F(2) duroc × pietrain pig resource population using different least-squares analysis models. Frontiers in Genetics, 2011, 2: 18.
[52] KIM J J, ZHAO H H, THOMSEN H, ROTHSCHILD M F, DEKKERS J C M. Combined line-cross and half-sib QTL analysis of crosses between outbred lines. Genetical Research, 2005, 85(3): 235-248.
[53] SESSA W C. The nitric oxide synthase family of proteins. Journal of Vascular Research, 1994, 31(3): 131-143.
[54] HATTORI Y, HATTORI S, KASAI K. Lipopolysaccharide activates Akt in vascular smooth muscle cells resulting in induction of inducible nitric oxide synthase through nuclear factor-kappa B activation. European Journal of Pharmacology, 2003, 481(2/3): 153-158.
[55] SILVA G B, GARVIN J L. Akt1 mediates purinergic-dependent NOS3 activation in thick ascending limbs. American Journal of Physiology Renal Physiology, 2009, 297(3): F646-F652.
[56] KOEBKE K J, PAULY D J, LERNER L, LIU X E, PACHECO A A. Does the oxidation of nitric oxide by oxyMyoglobin share an intermediate with the metMyoglobin-catalyzed isomerization of peroxynitrite? Inorganic Chemistry, 2013, 52(13): 7623-7632.
[57] LOKE K E, LAYCOCK S K, MITAL S, WOLIN M S, BERNSTEIN R, OZ M, ADDONIZIO L, KALEY G, HINTZE T H. Nitric oxide modulates mitochondrial respiration in failing human heart. Circulation, 1999, 100(12): 1291-1297.
[58] RAYNER B S, HUA S S, SABARETNAM T, WITTING P K. Nitric oxide stimulates myoglobin gene and protein expression in vascular smooth muscle. The Biochemical Journal, 2009, 423(2): 169-177.
Integrated Aanalysis of Genome and DNA Methylation for Screening Key Genes Related to Pork Quality Traits
ZHAO ZhenJian, WANG Kai, CHEN Dong, SHEN Qi, YU Yang, CUI ShengDi, WANG JunGe, CHEN ZiYang, YU ShiXin, CHEN JiaMiao, WANG XiangFeng, TANG GuoQing
College of Animal Science and Technology, Sichuan Agricultural University/Key Laboratory of Livestock and Poultry Multi-Omics, Ministry of Agriculture and Rural Affairs/Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province/State Key Laboratory of Swine and Poultry Breeding Industry, Chengdu 611130
【Background】 The meat quality traits of pigs are important economic traits. Studying the molecular mechanisms that affect various meat quality traits and discovering key genes can guide genetic improvement of pigs and have significant implications for improving pork quality. Currently, those researches on meat-related mechanisms were mainly based on DNA genomics, while studies on DNA methylation related to meat quality traits, as well as integrated analysis of combining genomics and methylation, are scarce. 【Objective】 The potential key genes affecting pork meat quality were screened and identified by combined analysis of genome and DNA methylome, so as to provide a reference for the genetic improvement of pork quality. 【Method】 In this study, 28 meat quality traits of the ongissimus dorsi muscle of 140 Large White pigs were examined. Epigenome-wide association analysis (EWAS) and genome-wide association analysis (GWAS) were performed to identify CpG and SNP sites significantly associated with each trait. Subsequently, the conditional association analysis was performed by using SNPs as covariates on the significantly associated sites overlapping between GWAS and EWAS to further identify CpG sites with independent effects. Association analysis was then conducted to identify methylation quantitative trait loci (meQTL) by using the methylation levels of CpG sites as the dependent variable and SNPs as the independent variable. Finally, the cis-methylation quantitative trait loci (cis-meQTL) were used as instrumental variables for Mendelian randomization analysis to infer the causal relationship between cis-meQTL and phenotypes, while potential key genes at the loci were annotated and identified. 【Result】 (1) The significant associated sites were identified in the same genomic regions for the meat quality traits, namely yellowness value at slaughter 45 minutes postmortem (b45min), drip loss (DL), and docosahexaenoic acid (C22:6n-3), by both EWAS and GWAS. (2) After conditional association analysis, seven CpG sites for b45minand one CpG site for DL remained significant, while the three sites for C22:6n-3 were no longer significant after using SNPs as covariates, indicating that the significant associations of the seven CpG sites for b45minand one CpG site for DL identified by EWAS were not influenced by nearby significant SNPs. (3) A total of ten meQTL were identified for the seven CpG sites for b45minand one CpG site for DL, but the majority were trans-meQTL, with only one CpG site (SSC12:44 254 675 bp) identifying a cis-meQTL, suggesting that this site might be regulated by nearby SNPs. (4) Mendelian randomization analysis showed a causal relationship between the CpG site (SSC12:44 254 675 bp) and the b45minphenotype. (5) annotation of the locus revealed that the nearest gene to the CpG site (SSC12:44 254 675 bp) and its cis-meQTL was NOS2, and the CpG site was located within the NOS2 gene. 【Conclusion】Based on the integrated analysis of DNA methylation and genomics data, this study proposed that the NOS2 gene might be a key candidate gene for meat color traits. DNA methylation and SNP jointly regulated gene expression, thereby affecting the expression of genes related to meat color traits.
epigenome-wide association study (EWAS); genome-wide association study (GWAS); meat quality traits; DNA methylation; meQTL

10.3864/j.issn.0578-1752.2024.07.014
2023-10-18;
2023-12-31
四川省科技厅项目(2020YFN0024, 2021ZDZX0008, 2021YFYZ0030)、四川省猪创新团队项目(sccxtd-2022-08)
赵真坚,E-mail:530773281@qq.com。通信作者唐国庆,E-mail:tyq003@163.com
(责任编辑 林鉴非)
猜你喜欢
今日农业(2022年2期)2022-11-16 12:29:47
美食(2022年4期)2022-04-16 00:25:37
美食(2020年5期)2020-06-01 10:14:38
现代园艺(2017年21期)2018-01-03 06:41:32
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年2期)2015-02-06 02:00:48
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
沈阳医学院学报(2014年4期)2014-12-27 13:44:30
作文评点报·作文素材小学版(2014年34期)2014-09-23 12:00:45
