
Alkali-resistant NOx reduction over FeVO4/TiO2 catalysts via regulating the electron transfer between Fe and V
2024-04-05 02:28YunangDongPengluWangXiangyuLiuJiangDengAlingChenLupengHanDengsongZhang
Chinese Chemical Letters 2024年2期
Yunang Dong ,Penglu Wang ,Xiangyu Liu,Jiang Deng,Aling Chen,Lupeng Han,Dengsong Zhang
International Joint Laboratory of Catalytic Chemistry,State Key Laboratory of Advanced Special Steel,College of Sciences,Shanghai University,Shanghai 200444,China
Keywords: Air pollution control NOX reduction Selective catalytic reduction Alkali metals Electron transfer
ABSTRACT The presence of alkali metals in exhaust gas from stationary resources causes a grand challenge for the practical application of selective catalytic reduction (SCR) of NOx with NH3.Here,alkali-resistant NOx reduction has been successfully implemented via tailoring the electron transfer over Fe and V species on FeVO4/TiO2 catalysts.The strong interaction between Fe and V induced electron transfer from V to Fe and strengthened the adsorption and activation of NH3 and NO over active VOx sites.In the presence of K2O,the strong electron withdrawing effect of Fe offset the electron donating effect of K on the VOx species,thus protecting the active species VOx to maintain the NOx reduction ability.The enhanced adsorption and activation of NH3 allowed SCR reaction to proceed via E-R mechanism even after K2O poisoning.This work elucidated the electronic effects on the alkali metals resistance of traditional ferric vanadate SCR catalysts and provided a promising strategy to design SCR catalysts with superior alkali resistance.
The emissions of nitrogen oxides (NOx) from coal-fired power stations and steel plants can trigger a series of environmental issues [1-3].Selective catalytic reduction of NOxwith NH3(NH3-SCR) is the most effective technology to eliminate NOxcurrently[4,5].Due to the satisfied SCR performance,V2O5-WO3(MoO3)/TiO2catalysts have been widely applied in stationary sources [6-8].However,there still exist some drawbacks,such as the narrow operating temperature window (300-400 °C) and the over-oxidation of SO2[9,10].Furthermore,the presence of alkali metals in the exhaust gas can lead to severe deactivation of the catalysts [11-13].Therefore,it is quite urgent to enhance the activity and alkali resistance of V-based catalysts to broaden the application among NH3-SCR [14-16].
So far,the FeVO4based catalyst has aroused extensive research interests because of its excellent low temperature activity and SO2resistance [17-19].Liuetal.proposed that the well-dispersed FeVO4catalysts exhibited high activity and N2selectivity.They also revealed that the true active sites in FeVO4catalysts were VOxspecies [20].Muetal.constructed a wide range of Fe1-xVxOδcatalysts with various Fe to V ratios and found that the electroninduced effects between Fe and V together with the interaction between FeVO4and Fe2O3species would greatly enhance the SCR performance [21].However,after K2O poisoning,the activity of FeVO4would decreased dramatically because of the destruction of acidity and redox properties [22,23].Traditional strategies for improving the resistance to alkali metals could be divided into two routes: (1) Enhancing the surface acidity and (2) separating the active sites and the poisoning sites [24-27].Basically,alkali metals are kinds of electron donors and their poisoning effects on catalysts mainly rely on the electron impactviareducing the valence of the boned active metal elements,thus undermining the Lewis acidity and redox capacity of the catalysts.Hence,weakening the electronic effect of alkali metals on the active species plays a crucial role in the resistance promotion of alkali metals on SCR catalysts [28,29].Lietal.modified commercial V2O5-WO3/TiO2catalysts with Ce and Cu and verified that the interaction between V,Ce,and Cu increased the ratio of V5+/(V5++V4+),which contributed to the excellent K2O resistance of the catalysts [30].However,the effect of electronic interactions between Fe and V on the alkali resistance of catalysts as well as the specific electron effect of alkali metals on the active V species had still not been clearly defined.
In this work,the FeVO4based catalysts with strong or weak interactions between Fe and V have been fabricated to investigate the role of electronic effects on the alkali resistance of SCR catalysts.The FeVO4catalyst preparedviaimpregnation method (i-FeV/Ti) is characterized as tightly bounded Fe and V with strong interaction,which shows superior SCR performance and alkali resistance than the catalyst with weaker interaction between Fe and Vviaco-precipitation preparation approach (denoted as p-FeV/Ti).The strong electronic withdraw ability of Fe increases the content of V5+on the surface,thus leading to promoted Lewis acidity and redox ability,which is favorable for the adsorption and activation of NH3and NO.After K2O poisoning,the electron-absorbing effects of Fe have offset the electron-donating effects of K2O on active V sites,therefore protecting the active VOxspecies to express the superior catalytic performance of NOxreduction.Subsequently,the adsorbed and activated NH3on the surface could react with gaseous NO and O2following Eley-Rideal mechanism even in the presence of K2O.This work provides a deep understanding of the effects of electronic interactions on the alkali resistance of FeVO4catalysts and paves a promising way to further design SCR catalysts with improved alkali resistance.
The i-FeV/Ti catalyst was prepared through a traditional impregnation method.First,0.3000 g oxalic acid was dissolved in 50 mL deionized water,then 0.0684 g ammonium metavanadate was added and stirred for 0.5 h,followed by the addition of 0.2363 g Fe(NO3)3·9H2O and 1 g TiO2.After stirring for another 2 h,they were dried using a rotary evaporator at 50 °C and then calcined at 500 °C for 4 h with a ramping rate of 5 °C/min.The resulting samples were labeled as i-FeV/Ti.The p-FeV/Ti catalyst was prepared by a co-precipitation method and alkali poisoned samples were obtained by impregnating 1 wt% K2O onto fresh catalysts.All details can be found in the Supporting information.
SCR performance was tested through a fixed-bed quartz flow reactor with 0.3 g catalysts.The reactant gas was composed of 500 ppm NO,500 ppm NH3,5 vol% O2and the carrier gas N2.The total gas hourly space velocity (GHSV) was controlled to be 50,000 h-1.NOxconversion and N2selectivity were calculated by the following equations (Eqs.1 and 2):
[NOx] represented the total concentration of NO and NO2,while[NOx]in,[NOx]out,[NH3]in,[NH3]out,[N2O]outindicated the concentrations of the corresponding gas at the inlet and outlet,respectively.
More detailed experimental parameters and characterizations could be found in Supporting information.
The i-FeV/Ti catalyst was prepared by impregnation method,which was characterized as tightly bounded Fe and V.Fig.1 shows the NOxconversion and the N2selectivity of i-FeV/Ti,K-i-FeV/Ti,p-FeV/Ti,and K-p-FeV/Ti catalysts under a gaseous hourly space velocity (GHSV) of 50,000 h-1.The i-FeV/Ti catalyst exhibits a broad temperature window of 210-390 °C with NOxconversion over 90%and N2selectivity nearly 100%.By contrast,the NOxconversion of p-FeV/Ti is lower within 150-300 °C.After K2O poisoning,two catalysts show quite different resistance performance.The SCR performance of i-FeV/Ti declines slightly within the whole temperature region.In contrast,the NOxconversion of p-FeV/Ti decreases greatly from 96% to 31% at 300 °C and stays below 45% within 120-390 °C.We also adjusted the content of Fe and V in i-FeV/Ti and found that when the ratio of Fe to V was 1:1,the catalyst showed the best activity and alkali resistance (Fig.S1 in Supporting information).In addition,we also test the SCR performance of i-FeV/Ti and p-FeV/Ti in the copresence of 1% K and 3% Pb and found that the performance of K&Pb-i-FeV/Ti remains superior than that of K&Pb-p-FeV/Ti (Fig.S2 in Supporting information).Besides,the activation energy of i-FeV/Ti and p-FeV/Ti was also tested under high GHSV (200,000 h-1) with NOxconversion below 20%(Figs.S3 and S4 in Supporting information).It was indicated that the i-FeV/Ti catalyst shows lower activation energy (39.37 kJ/mol)than p-FeV/Ti (51.07 kJ/mol),demonstrating its higher intrinsic activity.Furthermore,after K2O poisoning,the activation energy of K-i-FeV/Ti (55.33 kJ/mol) remains lower than that of K-p-FeV/Ti(64.76 kJ/mol).The tightly bounded structure between Fe and V is more conducive to induce strong electronic interactions,that should be the reason why i-FeV/Ti exhibited superior low temperature activity and alkali resistance [20,31].

Fig.1.NOx conversion and N2 selectivity of i-FeV/Ti,K-i-FeV/Ti,p-FeV/Ti and K-p-FeV/Ti catalysts.Reaction conditions: 500 ppm of NO,500 ppm of NH3,5 vol% O2,N2 as the balance gas,GHSV of 50,000 h-1.
To probe the element distribution on the surface of these catalysts,the high-resolution transmission electron microscopy(HRTEM) and energy-dispersive X-ray spectroscopy (EDS) were carried out.The HRTEM image in Fig.2A shows that there are no large FeVO4crystalline particles on the TiO2support for i-FeV/Ti,indicating that the active components are more likely to present in an amorphous form.Besides,the Fe and V elements are uniformly dispersed on the i-FeV/Ti catalyst,as seen from the elemental distribution mapping results in Fig.2B.However,there are some aggregations of Fe species on p-FeV/Ti,exhibiting grain-like characteristics in Fig.2B.It could also be found that distribution tendencies of Fe and V are almost the same on the surface of i-FeV/Ti in Fig.2C,indicating that Fe element is closely combined with V species.However,the line sweep results for Fe on p-FeV/Ti catalysts show a wave-like trend in Fig.2C,further demonstrating the inhomogeneous distribution of Fe.The poor distribution of Fe leads to limited bindings with V species,resulting in weaker electron interactions between Fe and V on p-FeV/Ti catalyst,which is the main reason for the worse activity and alkali resistance.

Fig.2.(A) HRTEM image of i-FeV/Ti.(B) EDX mapping of i-FeV/Ti and p-FeV/Ti.(C) EDX Line sweep of i-FeV/Ti and p-FeV/Ti.(D) UV-vis spectra and (E) Raman spectra of i-FeV/Ti,K-i-FeV/Ti,p-FeV/Ti,and K-p-FeV/Ti catalysts.
In addition,UV-vis spectra were performed to further characterize the coordination structure of these catalysts.As shown in Fig.2D,the peaks at 234 and 311 nm are attributed to Fe3+←O charge transfer of isolated Fe ions in tetrahedral and octahedral coordination,respectively.While the d-d transition band located at 799 nm is the fingerprint of V4+[34,35].Additionally,p-FeV/Ti exhibits an obvious band at 501 nm which should be ascribed to Fe2O3nanoparticles [36,37].Similar bands are not observed on i-FeV/Ti,which is consistent with the EDX mapping and line sweep results.Moreover,theinsituUV-vis DRS spectra were also performed at 240 °C to verify the active species on i-FeV/Ti and p-FeV/Ti (Figs.S6 and S7 in Supporting information).After the introduction of NH3or NO+O2,the variations mainly occur in the range of 510-800 nm,corresponding to the d-d transition bands of V3+/V4+[38].As for the bands at 234,311 and 501 nm corresponding to the Fe sites,there is no obvious change after the flowing of reaction gasses.This result demonstrates that the real active sites in the FeVO4catalysts are polymeric VOxspecies,which is consistent with the reported literatures [20,39].
Raman spectra were also collected to further identify the structure of all catalysts in Fig.2E.All the spectra show the secondorder feature of TiO2at 795 cm-1[40].The band at 923 cm-1should be ascribed to V=O stretches at polymeric vanadyl species for i-FeV/Ti [6].As for p-FeV/Ti and K-p-FeV/Ti catalysts,the peaks corresponding to the polymeric V species are significantly weaker.Due to the overlay of TiO2peaks,the bands related to Fe species could not be observed.Therefore,to further exclude the influence of support on the observation of Fe and V species among i -FeV/Ti and p-FeV/Ti catalysts,we continued to prepare bulk i-FeV and p-FeV materials to perform the Raman spectroscopy.It can be observed that the bands at 320,363,454,645,719,753,821,882,915,945 cm-1should be attributed to the representative FeVO4and bands at 212,272 cm-1are ascribed to hematite phase (Fig.S8 in Supporting information) [41].It is obvious that p-FeV shows much stronger characteristic of Fe2O3than i-FeV,and the peaks at 645 cm-1corresponding to the bridging Fe-O-V bands are more prominent on i-FeV,indicating tighter bonds between Fe and V[42].The Raman spectra and EDS results demonstrate that i-FeV/Ti catalysts possessed more Fe-O-V sites than p-FeV/Ti catalysts.
To clarify the electronic interactions between Fe and V species,X-ray photoelectron spectroscopy (XPS) was performed to probe the valence states of elements on the catalysts surface.As shown in Fig.3A,the V 2p3/2characteristic peak of i-FeV/Ti is centered at 517.39 eV [43].Compared with p-FeV/Ti (517.19 eV),V 2p peaks of i-FeV/Ti show higher binding energies.This variation can be caused by the transfer of more electrons from V to Fe.After K poisoning,the location of V 2p3/2characteristic peak on K-i-FeV/Ti barely changes but that on K-p-FeV/Ti shifts to the lower binding energy,indicating that the introduction of K2O reduces the valence states of V element on the surface for p-FeV/Ti but barely effect i-FeV/Ti.Besides,the V5+/(V5++V4+) ratio of i-FeV/Ti (89.31%) is higher than that of p-FeV/Ti (86.48%),which is in favor of NH3adsorption due to the stronger Lewis acidity [44].Furthermore,the V5+/(V5++V4+)ratio slightly changes after K2O is introduced (88.24%) for i-FeV/Ti.As for p-FeV/Ti,the ratio decreases from 86.48 to 81.84% after K2O poisoning.This phenomenon indicates thatK+imposes less influence on the electronic property of VOxspecies of i-FeV/Ti [39].The ratios of Fe3+/(Fe3++Fe2+) were also calculated based on the Fe 2p XPS results shown in Fig.3B.There are less Fe3+on i-FeV/Ti(57.91%) than on p-FeV/Ti (63.57%).The different ratio of Fe3+on two samples exactly proves the electron transfer from V to Fe over i-FeV/Ti,which has mentioned above.When K2O is introduced,the amount of Fe3+declined from 57.91% to 51.03% in i-FeV/Ti.As for p-FeV/Ti,it changes slightly from 63.57% to 60.26%.For K 2p XPS,the peaks located on the same place on both i-FeV/Ti and p-FeV/Ti(Fig.S9 in Supporting information).K2O is a kind of electron donor that would damage the redox capacity of SCR catalysts,but the electron withdrawing function of Fe protect the true active sites of VOxspecies from the attacking of K2O [33].As for the O 1s XPS spectra of all catalysts (Fig.S10 in Supporting information),the peaks located at~531.46 and~530.16 eV are attributed to the surface absorbed oxygen (denoted as Oα) and the lattice oxygen(denoted as Oβ),respectively [45].Generally,Oαis easier to be involved in the SCR reaction [46].The Oα/(Oα+Oβ) ratios of i-FeV/Ti,K-i-FeV/Ti,p-FeV/Ti,K-p-FeV/Ti are 19.98%,15.99%,12.68%,and 7.61%,respectively.The higher amounts of Oαon the surface of i-FeV/Ti can be resulted from the more O vacancies on the surface of i-FeV/Ti,which may be resulted from the strong interaction between Fe and V species.The introduction of K2O would certainly cause the loss of O vacancies,leading to the decline of Oaratios [22].While,the loss of O vacancies among K-i-FeV/Ti is less than that of K-p-FeV/Ti,further confirming the strong interaction between Fe and V species as well as the electron withdrawing function of Fe protect the active VOxsites from K2O poisoning.

Fig.3.(A) XPS spectra of V 2p and (B) Fe 2p of i-FeV/Ti,K-i-FeV/Ti,p-FeV/Ti and K-p-FeV/Ti catalysts.(C,D) H2 temperature-programmed reduction (H2-TPR) profiles and(E) NH3-TPD-MS profiles of i-FeV/Ti,K-i-FeV/Ti,p-FeV/Ti,and K-p-FeV/Ti catalysts.(F) In situ DRIFTS of NH3 desorption of i-FeV/Ti and p-FeV/Ti catalysts.
In NH3-SCR reaction,the acidity and redox properties of the catalyst are critical to its performance.The redox ability of catalysts was tested by H2temperature-programmed reduction (H2-TPR).As illustrated in Fig.3C,the reduction peaks at 346,372,and 387 °C for the i-FeV/Ti catalyst should be assigned to the reduction of Fe3+to Fe(3-δ)+,Fe(3-δ)+to Fe2+,and V5+to V4+,respectively [22].After K2O poisoning,these peaks migrate to higher temperature and the reduction peak of Fe3+to Fe(3-δ)+,Fe(3-δ)+to Fe2+overlap,indicating that the redox capacity of catalysts are affected.Besides,the peak attributed to V5+to V4+shifts from 387 °C to 414 °C after K2O poisoning.Compared with p-FeV/Ti,it can be found that the redox capacity of V species on i-FeV/Ti is less affected.For p-FeV/Ti shown in Fig.3D,the peaks located at 543 °C should be ascribed to V4+to V3+,which is not apparent on i-FeV/Ti,further evidencing the presence of more V4+on the surface of p-FeV/Ti [47].Furthermore,this peak becomes more pronounced after K2O poisoning,indicating that K2O poisoning exerts a greater influence on the V species of p-FeV/Ti.Besides,the H2consumption of i-FeV/Ti (1.252 mmol/g) and Ki-FeV/Ti (1.375 mmol/g) are higher than p-FeV/Ti (0.909 mmol/g)and K-p-FeV/Ti (0.986 mmol/g),further proving the superior redox ability of i-FeV/Ti.
In addition to redox capacity,the acidity is also an important factor affecting the performance of SCR catalysts.Therefore,the NH3temperature-programmed desorption mass spectrometry(NH3-TPD-MS) was undertaken to examine the acidity of all catalysts.As shown in Fig.3E,the peak at 191 °C assigned to weak acid,the peak at 238 °C attributed to medium strong acid,and the other one at 318 °C presenting strong acid are observed on i-FeV/Ti.The NH3desorption amount of i-FeV/Ti (19.19 μmol/g) is much higher than that of p-FeV/Ti (12.19 μmol/g).Actually,the NH3absorption ability of Fe species is quite poor [48],but the strong electronwithdrawing property enhances the acidity of VOxspecies,which endows i-FeV/Ti with better NH3absorption.After K2O poisoning,The NH3desorption amount of K-i-FeV/Ti (7.38 μmol/g) remains higher than that of K-p-FeV/Ti (5.21 μmol/g).Insitudiffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of NH3desorption was also used to analyze the adsorption and desorption behavior of NH3as well as the specific desorbed NH3species in Fig.3F.The bands at 1665,1437 cm-1are ascribed to NH4+absorbed on Brønsted acid sites,and bands located at 1602,1235,1183 cm-1are attributed to NH3absorbed on Lewis acid sites [49-55].It can be observed that the intensity of the desorption peaks of i-FeV/Ti are significantly higher than those of p-FeV/Ti.These results demonstrate that i-FeV/Ti possess much stronger surface acidity than that of p-FeV/Ti.When the K2O was corporate,the adsorbed species as well as the peak intensities on i-FeV/Ti barely change at low temperatures (<200 °C) (Fig.S11 in Supporting information).As for p-FeV/Ti,there are dramatic reductions in peak intensities (Fig.S12 in Supporting information).The effect of K2O on the surface acidity can be divided into two aspects: (1) occupying the Brønsted acid sites and (2) reducing the elemental valence thus weakening the Lewis acid strength through electronic effects.Therefore,due to the offset effect of the electron interactions caused by strong interaction of Fe and V on i-FeV/Ti,the Lewis acidity of active VOxspecies is maintained after K2O poisoning.
Furthermore,insituDRIFTS were also displayed to clarify the NO adsorption behaviors over fresh and K2O-poisoned catalysts.For i-FeV/Ti,the bands at 1625,1577,1364 and 1294 cm-1are attributed to bridged nitrate,bidentate nitrate,cis-N2O22-and monodentate nitrate,respectively (Fig.S13 in Supporting information)[50,51,53,55].As the temperature raised,the peaks at 1625 and 1577 cm-1gradually decline and those at 1364 cm-1increase,indicating that bridged nitrate and bidentate nitrate are progressively transformed tocis-N2O22-.It is reported thatcis-N2O22-generated on the surface would greatly enhance the NO conversion [56].That should be one reason why i-FeV/Ti exhibits superior SCR activity.As for p-FeV/Ti,the peaks at 1241 and 1623 cm-1ascribed to bridged nitrate are more stable (Fig.S14 in Supporting information) [51,52].Compared with i-FeV/Ti,the peaks attributed tocis-N2O22-are weaker and appear at higher temperature,demonstrating its inferior redox ability.What is more,for p-FeV/Ti,K2O addition promoted the formation of thermally stable absorbed NOxspecies,which would cover the active sites,including bridged nitrate (1605 cm-1),bidentate nitrate (1576 cm-1),and monodentate nitrate (1300 cm-1),leading to inferior K2O-resistance (Fig.S15 in Supporting information) [57,58].However,the intensities of absorbed nitrate on i-FeV/Ti are significantly reduced after K2O poisoning (Fig.S16 in Supporting information).The negative effect of K2O is greatly alleviated by the strong electronic interaction between Fe and V over i-FeV/Ti.
In order to clarify the whole reaction pathways over all catalysts,insituDRIFTs of the transient reaction between NO+O2and preabsorbed NH3species were carried out at 240 °C,at which the activity difference between K-i-FeV/Ti and K-p-FeV/Ti is greatest.After NH3is absorbed on i-FeV/Ti for an hour,the surface absorbed ammonia species mainly include ionic NH4+(1421 cm-1),coordinated NH3(1601,1255 cm-1) and -NH2amide species (1326 cm-1) (Fig.S17 in Supporting information) [53,59-61].They are consumed in 2 min with the addition of NO+O2.Then the peaks ascribed to bridged nitrate (1619 cm-1),bidentate nitrate (1256 cm-1) andcis-N2O22-(1376 cm-1) appear [57,62,63].It is obvious that the absorbed ammonia species would easily participate in the SCR reaction.As for p-FeV/Ti,the peak intensities of ammonia species absorbed on the surface,which should be attributed to NH3(1602,1243,and 1204 cm-1) on the Lewis acid sites,are relatively lower,proving its inferior acidity (Fig.S18 in Supporting information) [51,52,55].For the transient reaction at 240 °C between NH3and preadsorbed NO+O2in Fig.4A,cis-N2O22-(1376 cm-1) and bridged nitrate (1623,1216 cm-1) are formed on i-FeV/Ti after adsorption of NO+O2[51,57,63].After the introduction of NH3,these peaks vanish within two minutes and the bands belong to NH4+(1421 cm-1),NH3(1605,1252 cm-1)and -NH2amide species (1329 cm-1) appear,proving that these nitrates possess high reactivity with NH3[53,55,60,61].For p-FeV/Ti in Fig.4B,after injecting NO+O2for 1 h,the peaks attributed to bridged nitrate (1619 cm-1),bidentate nitrate (1558 cm-1),cis-N2O22-(1362 cm-1),and monodentate nitrate (1291 cm-1) form and these adsorbed nitrate species could contributed to the SCR reaction [50,53,61].However,the intensity of these peaks is quite lower than those observed in i-FeV/Ti,indicating its inferior redox ability.According to the above phenomena,we could find that the preabsorbed NH3species could react with NO+O2and the preabsorbed NOxspecies could also react with NH3on these fresh catalysts,demonstrating that the reactions on the surface would both follow Langmuir-Hinshelwood mechanism and Eley-Rideal mechanism.
The large amber windows were open, and the fish swam in, just as the swallows fly into our houses when we open the windows, excepting that the fishes swam up to the princesses, ate out of their hands, and allowed themselves to be stroked
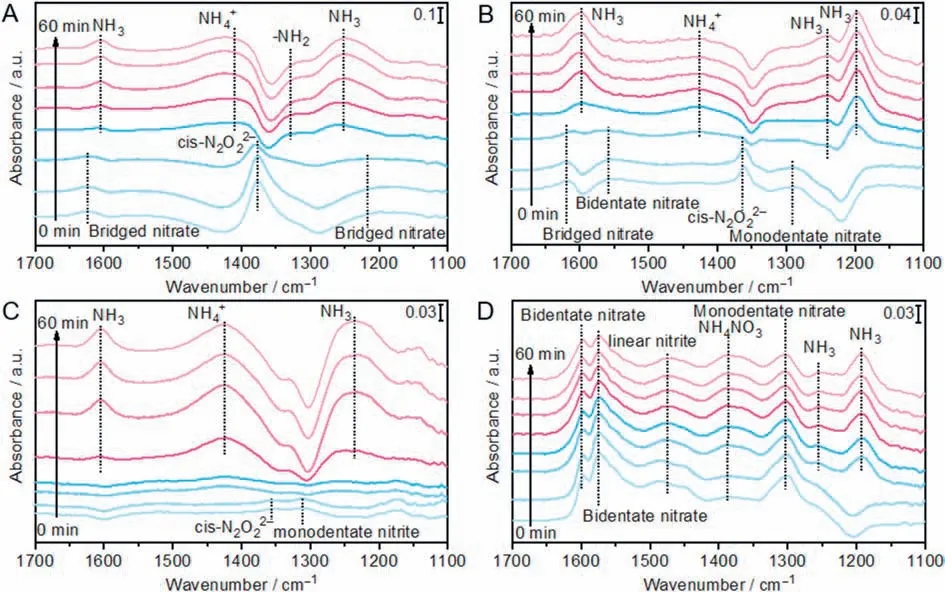
Fig.4.In situ DRIFTS of the transient reactions at 240 °C between NH3 and preadsorbed NO+O2 of (A) i-FeV/Ti,(B) p-FeV/Ti,(C) K-i-FeV(p)/Ti,and (D) K-p-FeV/Ti catalysts.
To elucidate the effect of K2O addition on the reaction pathway,insituDRIFTs of the transient reactions were also carried out on K2O poisoned catalysts.Compared with fresh i-FeV/Ti,similar species such as NH3(1605,1240 cm-1),NH4+(1425 cm-1) and-NH2(1332 cm-1) form on K-i-FeV/Ti after the injection of NH3for 1 h (Fig.S19 in Supporting information),but the intensities of these bands are reduced,which indicate that the addition of K2O suppress the acidity on the surface [49,53,55,61].A similar phenomenon also occur over K-p-FeV/Ti,but the differences are that the peak intensities are quite lower on K-p-FeV/Ti.After the introduction of NO+O2,the absorbed NH3species are quickly consumed and the subsequent peaks attributed to absorbed NOxspecies are quite weak on i-FeV/Ti,while a large amount of inactive nitrate species such as bidentate nitrate (1597,1574 cm-1),linear nitrite(1484 cm-1) and monodentate nitrate (1300 cm-1) accumulate on K-p-FeV/Ti (Fig.S20 in Supporting information) [52,55,60,61].Similar tendency also occur on the transient reaction at 240 °C between NH3and preadsorbed NO+O2.After preadsorption of NO+O2for 1 h,only quite few amounts of NOxspecies such ascis-N2O22-(1356 cm-1),monodentate nitrite (1311 cm-1) and chelating nitrite(1174 cm-1) are preserved on i-FeV/Ti in Fig.4C.With the addition of NH3,they are consumed in 5 min and corresponding NH3/NH4+species form on the surface [50,55,58].In contrast,after the flowing of NO+O2for 1 h,a large number of stable nitrate species cover the surface of p-FeV/Ti in Fig.4D.It was reported that K would not affect the formation of ammonium nitrate species,but it would restrain the decomposition of ammonium nitrate species to N2and H2O [28].With the introduction of NH3,these peaks do not change at all while some bands (1254 cm-1,1193 cm-1) attributed to the absorbed NH3appear.It indicates that these stable nitrates cover the active V sites and hinder the process of SCR reaction on K-p-FeV/Ti,while on K-i-FeV/Ti,the strong electron interaction between Fe and V protect the active V species and promote SCR reaction [55,60].Based on these results,after K poisoning,the reactions occur on i-FeV/Ti mainly follow the Eley-Rideal mechanism while the Langmuir-Hinshelwood mechanism is weakened,which is similar with other K poisoned V-based catalysts [11].
In summary,the effects of electron interactions on alkali resistance over FeVO4/TiO2for NH3-SCR are revealed.From Fig.5,it can be found that due to the strong electron-withdrawing effect of Fe,the valence of V element on the surface is increased,which facilitates the adsorption and activation of NH3.After alkali poisoning,the electron-absorbing effect of Fe offset the electron-giving effect of K2O on V element,thus protecting the real active sites of VOxspecies.It allows the catalysts to participate in the SCR reaction through E-R mechanism even in the presence of K2O.This work provides a profound comprehension of the electron interactions effects on the alkali resistance of catalysts and paves a promising way for the design of highly alkali-resistant SCR catalysts in the future.
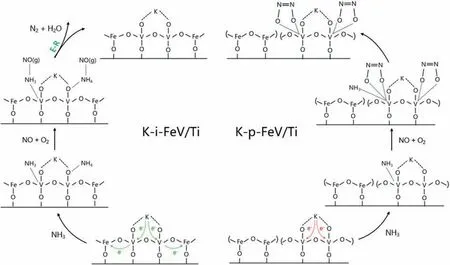
Fig.5.Possible reaction pathway of K poisoned i-FeV/Ti and p-FeV/Ti catalysts.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge the National Natural Science Foundation of China (No.22125604),Shanghai Rising-Star Program (No.22QA1403700) and Chenguang Program supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (No.22Z00354).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108635.
 Chinese Chemical Letters2024年2期
Chinese Chemical Letters2024年2期
- Chinese Chemical Letters的其它文章
- The 3rd Xihua Chemistry and Biomedicine Forum
- Professor Hualiang Jiang: A tribute to an esteemed visionary chemist and pharmacist
- Recent advances in visible light-mediated chemical transformations of enaminones
- Development of porphyrin-based fluorescent sensors and sensor arrays for saccharide recognition
- Recent advances of versatile reagents as controllable building blocks in organic synthesis
- Synthetic host-guest pairs as novel bioorthogonal tools for pre-targeting☆