
Recent advances of versatile reagents as controllable building blocks in organic synthesis
2024-04-05 02:27ShaominChenLuigiVaccaroYanlongGu
Chinese Chemical Letters 2024年2期
Shaomin Chen ,Luigi Vaccaro ,Yanlong Gu
a Key Laboratory of Material Chemistry for Energy Conversion and Storage,Ministry of Education,Hubei Key Laboratory of Material Chemistry and Service Failure,School of Chemistry and Chemical Engineering,Huazhong University of Science and Technology,Wuhan 430074,China
b Laboratory of Green S.O.C.,Dipartimento di Chimica,Biologia e Biotecnologie,Università degli Studi di Perugia,Via Elce di Sotto 8,Perugia 06123,Italy
c School of Chemistry and Chemical Engineering,The Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan,Shihezi University,Shihezi 832004,China
Keywords: Versatile reagents Divergent synthesis Dual synthons Bond cleavage Tandem reactions
ABSTRACT Diversity-oriented synthesis is a powerful and interesting synthetic tool for the rapid construction of structurally complex and privileged scaffolds from readily accessible starting materials.To date,diversityoriented synthesis mostly relies on the employment of versatile reagents.Versatile reagents can be regulated as controllable and flexible building blocks for multipurpose utilizations.Over the past decade,a variety of multifunctional reagents have been developed.However,most versatile reagents usually need multi-step synthesis,thus restricting their wide application to a large extent.In terms of the practicalities and universalities,we prefer to pay more attention to the utilization of simple and practical versatile reagents with multiple reactivities,mainly including atropaldehyde acetals,aryl methyl ketones,vinylene carbonate,vinyl azides,aryldiazonium salts,rongalite,halodifluoromethyl compounds.Most importantly,these versatile reagents can also play different roles simultaneously in the same reaction,in which their different reactivities are converged into the final target products.Such strategy can not only offer more possibilities for the synthesis of several active pharmaceutical ingredients,but also minimize the occurrence of some side reactions by lessening the varieties of materials.Also,a perspective is given at the end of this review.
1.Introduction
Diversity-oriented synthesis has emerged as a powerful and useful synthetic tool for the construction of a wide range of structurally complex privileged scaffolds from readily accessible starting substrates,which has attracted considerable attention over the past decade [1-4].Currently,diversity-oriented synthesis mostly relies on the utilization of versatile reagents [5].Generally,versatile reagents refer to the class of highly energetic agents with multiple reactivities,which can be regulated for multipurpose utilizations.Therefore,it is promising to generate structurally diverse molecules by combining versatile reagents with various counterparts under the certain reaction conditions (Fig.1).In recent years,some common and simple molecules,such as dimethyl sulfoxide(DMSO) [6,7],N,N-dimethylformamide (DMF) [8],and dimethyl carbonate (DMC) [9-13],as well as isocyanides [14-16],have been put forward as flexible and versatile building blocks for organic synthesis.With the increasing demand for a large number of functionalized molecules,developing novel versatile reagents and enriching their reactivities are of great significance and appealingly needed.
Owing to the elusive reactivities of these versatile reagents,how to finely regulate their reactivities and overcome the intrinsic bias,and to develop new and specific bond-forming modes,is the major intractable problem [17,18].If possible,converging their different reactivities together based on the design of multicomponent reaction pathways should be also taken into consideration,since such strategies can not only offer more possibilities for the synthesis of diverse and complex products,but also advantageously minimize the occurrence of side reactions by lessening the varieties of materials.Apparently,there are two formidable challenges in such transformations,which includes (i) finding suitable counterparts to couple with these energetic reagents in a welldesigned sequence;and (ii) identifying compatible catalytic systems to bind all of the individual reaction steps,perhaps due to the existence of the big differences between each catalytic cycle.Therefore,continuous efforts from a large number of research groups need to be devoted to this field.
Although novel multifunctional reagents are emerging in numerous research articles,to the best of our knowledge,there has been no systematic review to summarize the recent advances of versatile reagents in organic synthesis so far.Meanwhile,it should be noted that most versatile reagents usually need multi-step synthesis,thus restricting their wide application to a large extent.To comprehensively acquaint with the current development of this realm,in this review,we prefer to focus on the utilization of simple and practical versatile reagents as flexible building blocks that have been investigated by numerous research groups for accessing value-added organic molecules.These reagents include atropaldehyde acetals,aryl methyl ketones,vinylene carbonate,vinyl azides,aryldiazonium salts,rongalite,halodifluoromethyl compounds (Fig.2).Furthermore,this review also highlights the reaction pathways that these diverse building blocks are generated under the inducement of different conditions.Most importantly,the integration of different reactivities of these versatile reagents into the target products can be also presented in these examples.

Fig.2.The emerging versatile reagents in organic transformations.
2.Atropaldehyde acetals
Atropaldehydes and their acetals are an important sort of highly energetic reagents,which can be readily prepared from styrenes orα-hydroxyacetophenones [19,20].Owing to the existence of an aldehyde group and C-C double bond,they can be employed as model 1,3-biselectrophiles under acidic conditions to undergo diverse annulations [21-23].In some cases,atropaldehydes acetals are also used as masked C2 donorsviathe cleavage of C-C bond,acting as the surrogates of phenylacetyl or styryl fragment(Scheme 1) [24].Intriguingly,atropaldehyde acetals can also perform the dual reactivities in some tandem reactions,playing a dual role of C2 and C3 synthons [17].These prominent properties successfully enable atropaldehyde acetals to provide divergent structural units for the construction of more privileged scaffolds.
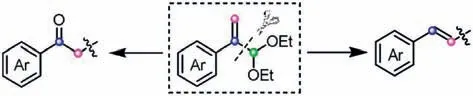
Scheme 1.Atropaldehyde acetals as masked C2 donors via the cleavage of C-C bond.
In 2018,our group firstly reported atropaldehyde acetals as the C3 reagents to couple with different bisnucleophiles,constructing diverse six-membered aromatic rings under the catalysis of CuBr2/Al(OTf)3or NBS/Al(OTf)3with high efficiencies (Scheme 2)[23].It should be mentioned that CuBr2or NBS played a vital role in promoting the reaction,in which CuBr2or NBS could enable the occurrence of the last aromatization step by a successive bromination/dehydrobromination,thus driving the reaction equilibrium towards final products.When 1,3-dicarbonyl compounds were engaged as bisnucleophiles,the salicylate products were obtained in moderate to excellent yields.Instead,the corresponding carbazole derivatives could be generated once employingα-(indol-2-yl) acetates as the counterparts.In addition,other N,C-and N,N-based bisnucleophiles were also compatible with this protocol,delivering various heterocyclic compounds smoothly.It is worth noting that this method can be also applied in the synthesis of antiinflammatory agent diflunisal,not only enhancing the yield,but also avoiding the high-pressure operation.
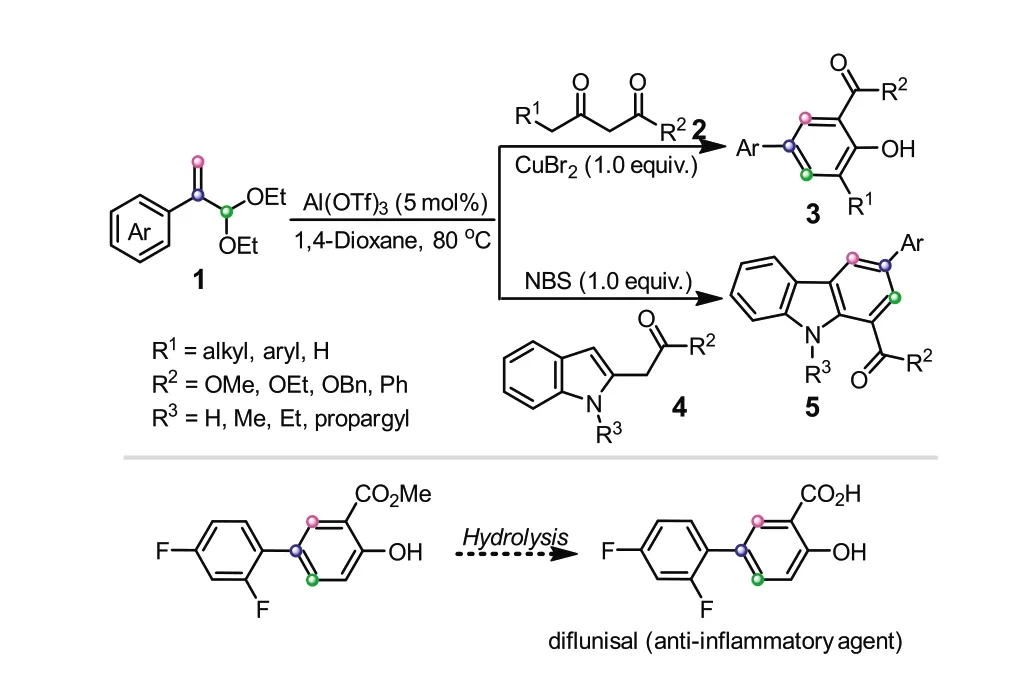
Scheme 2.Atropaldehyde acetals as C3 donors for the construction of sixmembered aromatic rings.
In addition,atropaldehyde acetals were further exploited as elegant C3 building blocks for the preparation of cyclohepta[b]indoles[25],which are an important class of indole-fused tricyclic structural framework and widely present in several alkaloids and active pharmaceutical ingredients.As demonstrated in Scheme 3,atropaldehyde acetals could capture the intermediate products 3,3′-bis(indolyl)methanes (BIMs) generated from indoles and 3,3-diethoxypropionates and undergo a [4+3] cycloaddition to deliver the target products with the assistance of PTSA in the green solvent anisole.Actually,BIMs served as 1,4-bisnucleophiles in the process,which can beinsitutransformed into 3-vinylindolesviathe removal of one molecule of indole.Also,the environmentalfriendly catalytic system was equally applicable to the threecomponent reaction of indoles,acetophenones and atropaldehyde acetals,giving the diaryl substituted cyclohepta[b]indoles in moderate yields.The concise and efficient strategy represented an alternative approach to accessing cyclohepta[b]indole compounds.

Scheme 3.Atropaldehyde acetals as C3 donors for the construction of cyclohepta[b]indoles.
In subsequent studies,we unexpectedly found that atropaldehyde acetals could serve as C2 fragments to incorporate into final products.In 2021,we reported this preliminary result,in which Lewis acid-catalyzed cascade reactions of atropaldehyde acetals were established,allowing the preparation of two important molecules,2,2-disubstituted indolin-3-ones and naphthofurans (Scheme 4) [24].For the synthesis of 2,2-disubstituted indolin-3-ones,2-arylindole firstly underwent a Michael addition with atropaldehyde acetal to result in the intermediate13.After that,the intermediate13was converted into the intermediate16through two different oxidation sequences under oxygen atmosphere,which both involved a crucial step,the oxidative cleavage of C-C bond,ensuring the formation of the phenylacetyl segment.Finally,the immigration of phenylacetyl occurred to furnish indolinone product.In a similar manner,the naphthofuran products were also afforded usingβ-naphthol to trap the C2 electrophilic species generated from atropaldehyde acetalsviathe C-C cleavage assisted by oxygen from the air (Scheme 5).

Scheme 4.Atropaldehyde acetals as C2 electrophiles for the synthesis of 2,2-disubstituted indolin-3-ones.
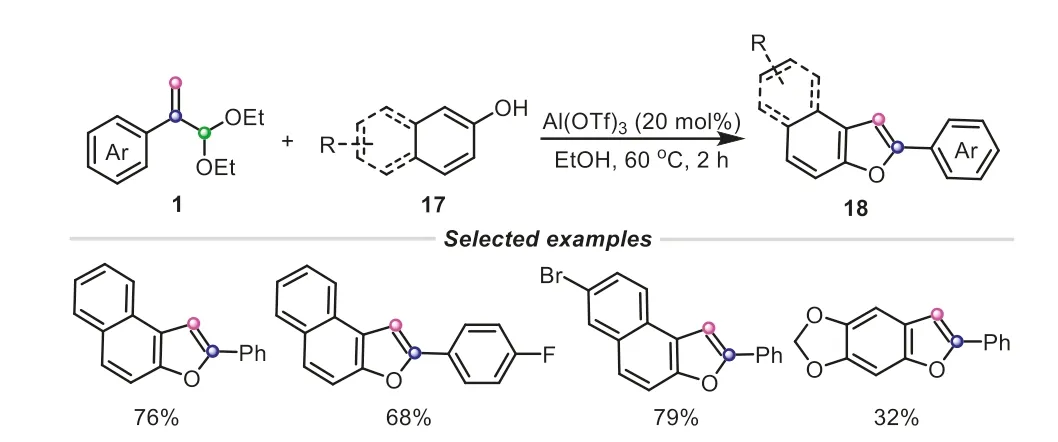
Scheme 5.Atropaldehyde acetals as C2 electrophiles for the synthesis of naphthofurans.
Intriguingly,the fracture mode of atropaldehyde acetals can be subtly regulated by the substrates.Using 1,3,5-trimethoxybenzene instead of 2-arylindole andβ-naphthol,to couple with atropaldehyde acetals,an intriguing C-C cleavage mode enabled by retro-Aldol reaction,might appear in this process,since neither oxygen nor nitrogen atmosphere could enhance the yield of product (Scheme 6) [24].Likewise,a successive Michael addition and dealcoholization process firstly occurred,forming an intermediate22,which then went through a tautomerization to generate the intermediate23.After that,the terminal carbon of23was attacked by another molecule of19and led to the carboncarbon bond scission,obtaining the final alkenylation product20.It should be noted that the retro-Aldol-induced C-C cleavage mechanism was further verified by the detection of by-product bis(2,4,6-trimethoxyphenyl)methane.
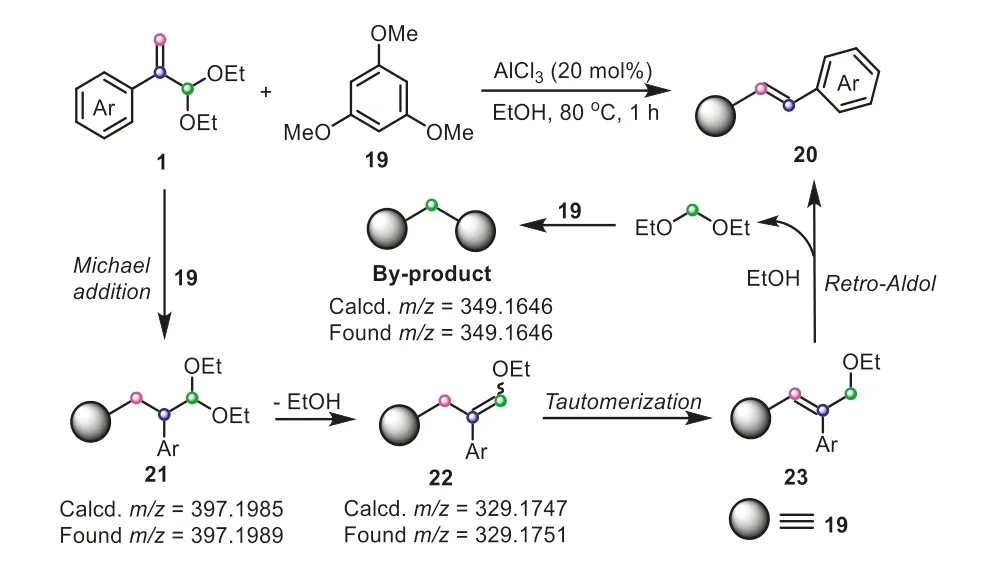
Scheme 6.Atropaldehyde acetals as C2 electrophiles for the synthesis of stilbene derivatives.
Inspired by these above results,we envisaged whether the dual reactivity of atropaldehyde acetals can be performed in the same reaction.To accomplish the goal,it was vital to find a suitable coupling partner.Then we attempted to utilizeN-substituted glycine esters to capture the electrophilic atropaldehyde acetals,because the reactive sites on glycine esters can be altered easily.Gratifyingly,in 2022,we disclosed a simple and novel method for the construction of pyrrolo[1,2-a]quinolines with atropaldehyde acetals andN-phenylglycine esters as the starting materials in the green medium diethyl carbonate (DEC) (Scheme 7) [17].The mechanistic studies suggested that the process started with a [3+2] cyclization to form the five-membered ring intermediate27.Owing to the inherent high nucleophilicity of phenyl in27,a SN2′ substitution with another molecule of atropaldehyde acetal quickly occurred,providing an intermediate28.Afterwards,the oxidative C-C cleavage provided a phenylacetyl moiety,enabling the subsequent occurrence of electrophilic cyclization to furnish the desired tricyclic product.In contrast with the conventional transition metal-catalyzed intramolecular cycloisomerizations to access this framework,this green and metal-free protocol successfully provided an expedient and efficient strategy to construct the structurally complex tricyclic skeleton in virtue of the dual functions of atropaldehyde acetals.
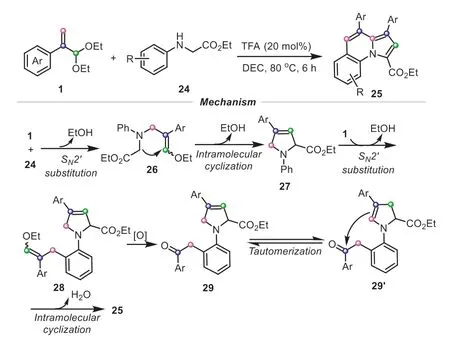
Scheme 7.Atropaldehyde acetals as dual C3/C2 synthons for the synthesis of pyrrolo[1,2-a]quinolines.
In addition toN-phenylglycine esters,N-benzylglycine esters could also well match with atropaldehyde acetals in the presence of TFA to offer 3,5-diarylpyridine compounds (Scheme 8) [17].Similarly,two molecules of atropaldehyde acetals participated in the process,in which atropaldehyde acetals played two roles,acting as a dual C3/C2 synthon.Different from the oxidative C-C cleavage in the last instance,the styryl fragment enabled by the retro-Aldol reaction was present in the transformation,which would smoothly render the formation of pyridine ring through the [3+2+1] cyclization.The above-mentioned transformations elegantly revealed the versatility of atropaldehyde acetals as multiple building blocks in organic synthesis.

Scheme 8.Atropaldehyde acetals as dual C3/C2 synthons for the synthesis of 3,5-diarylpyridines.
3.Aryl methyl ketones
Aryl methyl ketones constitute a type of common and inexpensive reagents in organic synthesis,and they have been extensively engaged as building blocks for the construction of valuable molecules.To date,aryl methyl ketones have been frequently served as functionalized C1 or C2 unit in many elegant works,due to the existence of electrophilic carbonyl and nucleophilicαmethyl.With the assistance of catalyst and additive,the umpolung ofα-methyl carbon may occur,thus rendering aryl methyl ketones to be biselectrophiles [26].In addition,it is also possible to lead to the C(CO)-C bond cleavage of ketones in some cases (Scheme 9).Overall,these rich reactivities confer much promise on aryl methyl ketones to become a class of versatile reagents.
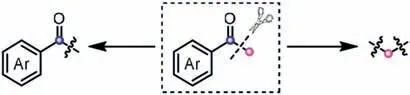
Scheme 9.Aryl methyl ketones as versatile synthons via the cleavage of C-C bond.
In 2018,our group developed an efficient and straightforward approach to synthesizing carbazole derivatives based on the threecomponent reaction of indoles,ketones andα-bromoacetaldehyde acetals in the presence of Bi(OTf)3(Scheme 10) [27].The aryl methyl ketone can provide an acetyl fragment to combine with tryptaldehyde-type acetal intermediate generated from indole andα-bromoacetaldehyde acetal,finally delivering the aryl substituted carbazole product.In addition,some aliphatic ketones,such cyclic ketone and 1,3-dicarbonyl compounds,were also suitable to this transformation,furnishing the corresponding carbazoles in satisfied yields.
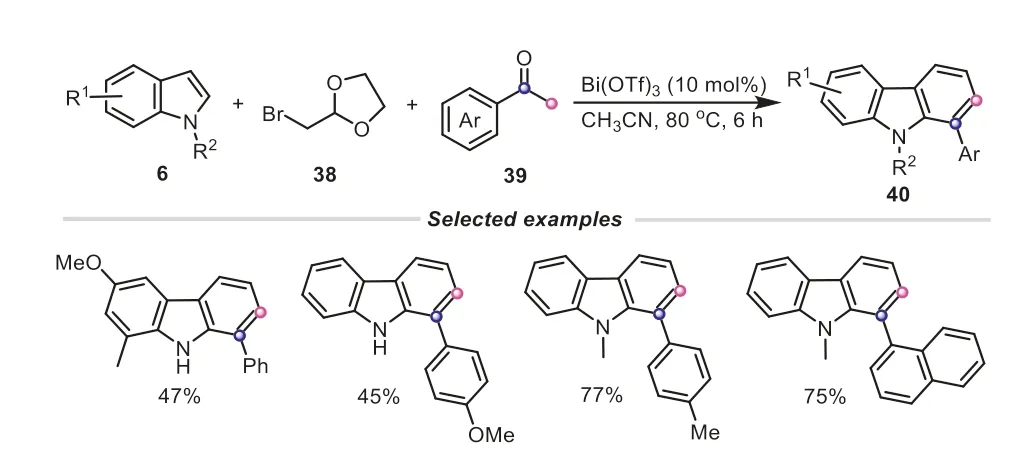
Scheme 10.Aryl methyl ketones as C2 synthons for the synthesis of carbazole derivatives.
Molecular iodine often serves as an additive to assist some transformations,especially for the aryl methyl ketones-participated reactions [26].For example,Chen’s group reported a cost-effective synthetic approach to obtain polysubstituted imidazoles from aryl methyl ketones and amidines under the assistance of ZnI2/I2(Scheme 11) [28].At the outset,this reaction went through an electrophilic activation of iodine,generating iodoacetophenone43.Afterwards,the nucleophilic substitution between amidine with43occurred and was followed by an intramolecular cyclization to afford the desired product.In addition,it is well-known that molecular iodine can promote the occurrence of Kornblum oxidation,rendering the conversion of aryl methyl ketones to highly reactive phenylglyoxal,which contributes to establishing cascade reactions for the synthesis of important molecules.In this respect,Wu’s group has made great progress.For instance,this group developed concise Lewis acid-catalyzed annulation strategies to obtain imidazoles and pyrrole-2-carbaldehydes in I2/DMSO (Scheme 12) [29,30].In these reactions,acetophenone initially went through a sequential iodination and Kornblum oxidation,forming the phenylglyoxal,which was subsequently captured by other reactive reagents to furnish the target products.

Scheme 11.Aryl methyl ketones as C2 synthons for the synthesis of polysubstituted imidazoles.
Scheme 12.Aryl methyl ketones as C2 synthons for the synthesis of imidazoles and pyrrole-2-carbaldehydes.
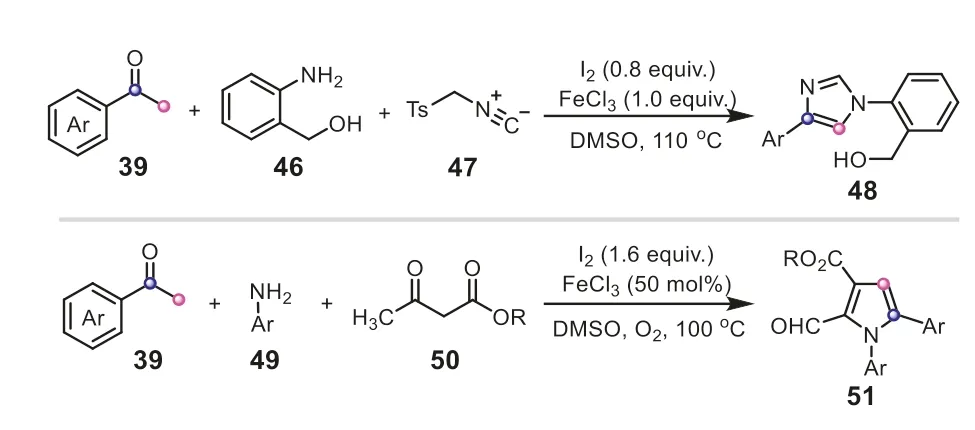
Apart from the common C2 units,aryl methyl ketones can act as C1 building blocks to construct diverse frameworks.The same group reported an expedient and highly efficient C(CO)-C bond cleavage of aryl methyl ketones for the preparation of 1,3,4-oxadiazoles (Scheme 13) [31].Similarly,the acetophenone compound was readily converted into the arylglyoxal54with the assistance of I2/DMSO through a sequential iodination and Kornblum oxidation.The aldehyde of54reacted with benzohydrazide to give the C-acyl benzoylhydrazone55.It was proposed that K2CO3-promoted oxidative iodination might be a crucial step,which subsequently rendered the selective cyclization of55(path a).Additionally,I2/K2CO3-assisted isomerization of55and cyclization was also possible to provide59(path b).Finally,59underwent a deacylation to offer the product53.It must be mentioned that the real-time monitor of13C labeling13C NMR spectroscopy also verified the reaction sequence that53was generated from59viathe cleavage of C(CO)-C(methyl) bond in aryl methyl ketones.In the subsequent investigation,the iodine-mediated unstrained C(sp2)-C(sp3) bond cleavage strategy of aryl methyl ketones was also applied to synthesize 1H-pyrazolo[3,4-b]pyridines (Scheme 14) [32].In this example,aryl methyl ketones equally served as a C1 synthon,merely providing a carbon atom to incorporate into the heterocyclic scaffold.These aforementioned results elegantly demonstrated the powerful functions of I2/DMSO system in assisting the conversion of aryl methyl ketone compounds.
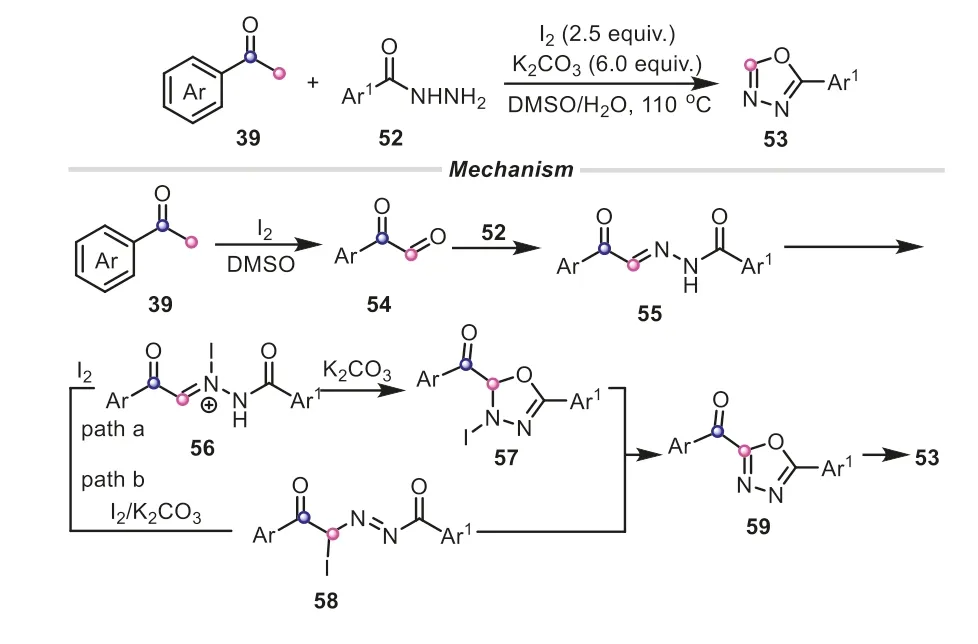
Scheme 13.Aryl methyl ketones as C1 synthons for the synthesis of 1,3,4-oxadiazoles.
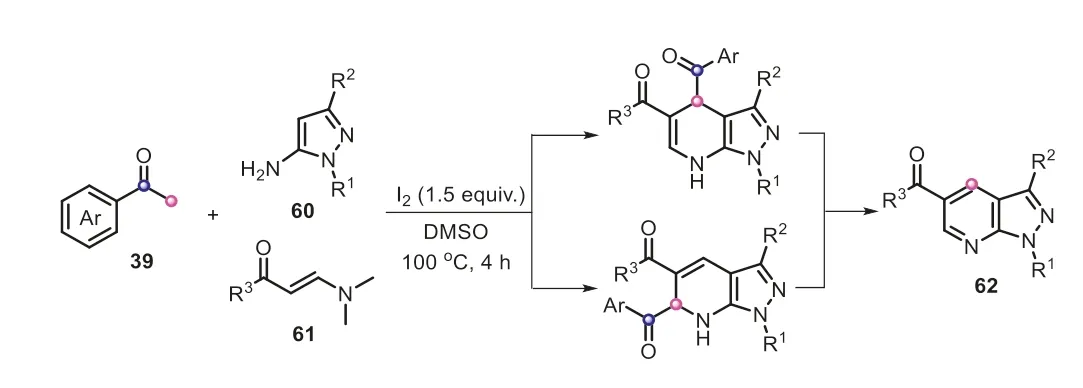
Scheme 14.Aryl methyl ketones as C1 synthons for the synthesis of 1Hpyrazolo[3,4-b]pyridines.
Benefiting from the scission of C(sp2)-C(sp3) bond,aryl methyl ketones can not only generate the smart C1 subunit,but also led to the benzoyl fragment.In the above-mentioned examples,we present the utilization of aryl methyl ketones as one carbon synthon in establishing the intermolecular annulation reactions.Next,we will demonstrate the application of the benzoyl fragment derived from aryl methyl ketones in organic synthesis.In 2014,Jiao’s group developed a straightforward conversion of aryl methyl ketones to amides by employing azide as the nitrogen source based on a copper-catalyzed aerobic oxidative cleavage of C(CO)-C(alkyl)bond (Scheme 15,left) [33].Several acetophenone derivatives,including some challenging aryl ketones bearing long-chain alkyl substituents,were all amenable to this transformation,giving the corresponding amides in generally good yields.In particular,some products can be regarded as the potential precursors of some important pharmaceutical agents.In the same year,a similar protocol was also proposed by the same group for the synthesis of esters from aryl methyl ketones (Scheme 15,right) [34].It should be noted that a wide range of alcohols,such as primary and secondary alcohols,as well as chiral alcohols,even phenols and various natural alcohols,were also compatible with the reaction.Considering that O2or air atmosphere was crucial to promoting this reaction,they performed some isotope experiments.It was found that 33%of carbonyl oxygen atom from the ester product was labeled under18O2atmosphere.In addition,the oxygen exchange of ester with water could not occur.Combined with the result that 10% of product was labeled upon conducting the reaction in the presence of H218O open to air,it was inferred that partial oxygen atom of ester carbonyl might come from the O2in air.Therefore,aryl methyl ketones merely provided the partial benzoyl fragment to deliver the product.

Scheme 15.Aryl methyl ketones as benzoyl fragments for the synthesis of amides and esters.
In light of the diverse reactivities of aryl methyl ketones performed in the aforementioned transformations,it is promising to assemble different reactivities together for the synthesis of organic functionalized molecules in some cases.For example,Wuetal.disclosed a novel and efficient I2-promoted formal [2+1+1+1]annulation for the construction of 1,2,5-trisubstituted imidazoles based on the multi-component reaction of aryl methyl ketones,anilines and tosylmethyl isocyanide (Scheme 16) [35],in which two molecules of aryl methyl ketones took part in the reaction,acting as C1 and C2 dual synthons.Firstly,phenylglyoxal wasin situgeneratedviaa successive iodination and Kornblum oxidation in the presence of DMSO.Then a part of reactive phenylglyoxal would react withp-toluidine to give the C-acylimine intermediate67,which was subsequently combined with amine derived from tosylmethyl isocyanide to afford the intermediate68viaa crosstrapping process.Eventually,the cyclocondensation of68with another molecule of phenylglyoxal successfully rendered the formation of the desired product66.It is apparent that aryl methyl ketones actually served as the roles ofα-dicarbonyl compounds and aldehydes to converge into the reaction.

Scheme 16.Aryl methyl ketones as dual C1/C2 synthons for the construction of 1,2,5-trisubstituted imidazoles.
4.Vinylene carbonate
Vinylene carbonate is a simple and widely used organic synthon featuring commercial availability and high operational security.Currently,vinylene carbonate can act as multiple roles in a plethora of organic transformations,such as becoming the surrogates of ethynol,acetylene and glyoxal,as well as an acetyl transfer reagent (Scheme 17).Hence vinylene carbonate has been regarded as an ideal synthetic precursor for divergent functionalization,especially in terms of the transition-metal-catalyzed C-H conversion by virtue of the extrusion of carbonate anion,leading to the generation of various value-added heterocyclic frameworks [36-38].In addition,different reactivities of vinylene carbonate can be concurrently converged into the target molecules in some cases,thus enriching the diversity and complexity of products.
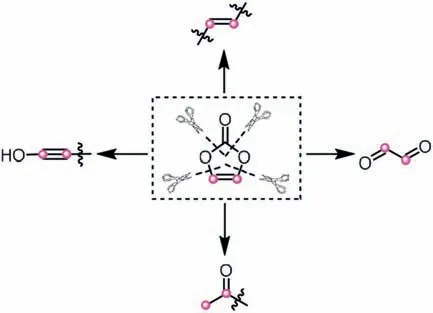
Scheme 17.Vinylene carbonate as versatile synthons via the cleavage of different C-O bonds.
In the early reports,it was deemed that vinylene carbonate can be a substitute of ethynolviadecarboxylation to furnish a variety of molecules.In 2009,Tanaka’s group firstly employed commercially available vinylene carbonate as an unstable ethynol surrogate to prepare substituted phenols through a decarboxylative[2+2+2] cycloaddition (Scheme 18) [39].The combination of 5 mol% of cationic rhodium(I) catalyst with BINAP ligand could efficiently render the occurrence of the reaction under mild conditions.At the outset of the reaction,diyne reacted with rhodium catalyst to produce rhodacyclopentadiene complex72.And the subsequent insertion of vinylene carbonate,followed by a reductive elimination of rhodium metal,generated carbonate73.With the assistance of rhodium(I),the elimination of CO2was facilitated to get the desired bicyclic phenol71.The reaction showed a wide scope towards diyne compounds,in which 1,6-diynes and 1,7-diynes with various substituent groups were well coupled with vinylene carbonate to deliver diverse bicyclic phenols in moderate to good yields.However,terminal 1,6-diyne and 1,7-diyne turned out to be inapplicable to the transformation,probably due to their rapid homo-[2+2+2] cycloaddition.
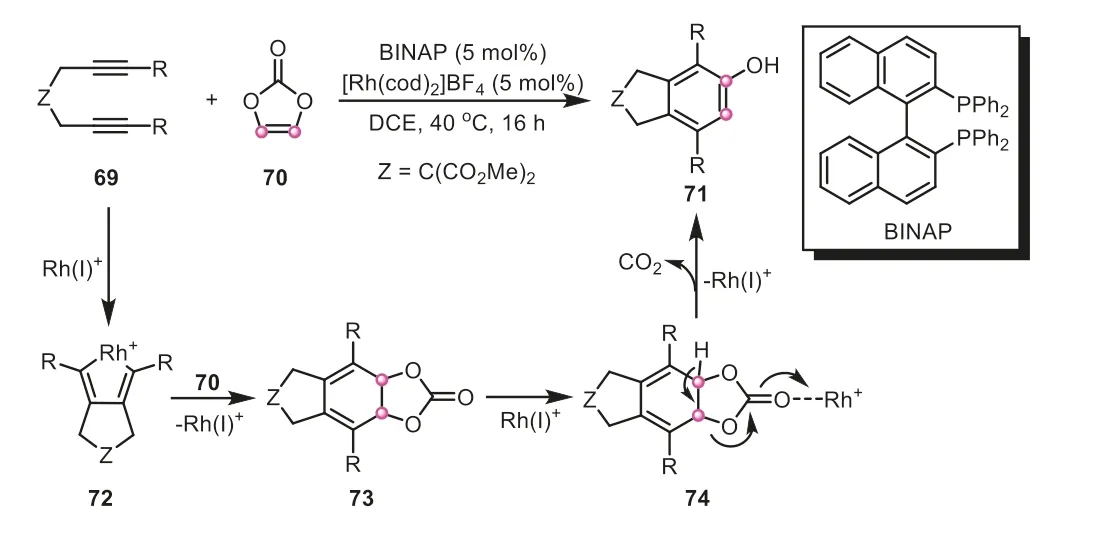
Scheme 18.Vinylene carbonate as ethynol equivalent for the synthesis of substituted phenols.
In 2019,Hayashi’s group applied the similar strategy for the synthesis of arylacetaldehydes [40].In the presence of iridium catalyst,vinylene carbonate successfully accomplished the formylmethylation of arylboronic acids,providing arylacetaldehydes in generally good yields.The catalytic cycle was proposed as shown in Scheme 19.An aryliridium species77generated by transmetalation was firstly combined with vinylene carbonate to give the intermediate78,which underwentβ-oxygen elimination to form 2-arylethenyl alcohol79.Subsequently,a successive hydrolysis/decarboxylation/keto-enol tautomerization process occurred,producing arylacetaldehyde.Interestingly,the hydrolysis of boronic acid to arene Ar-H was much slower without B2(OH)4.It was conjectured that B2(OH)4as a co-catalyst could accelerate the protodeboronation of ArB(OH)2,corresponding to the transmetalation and/or hydrolysis steps.In comparison with typical oxidation or reduction methods,this strategy offered a simple and alternative synthetic route to access the arylacetaldehydes.By utilizing the similar strategy,Wang and Zhouetal.exploited 2-arylquinazolinones to capture the ethynolinsitugenerated from vinylene carbonate to gain fused quinazolinones by ruthenium(II)-catalyzed C-C/C-N annulation (Scheme 20) [41].
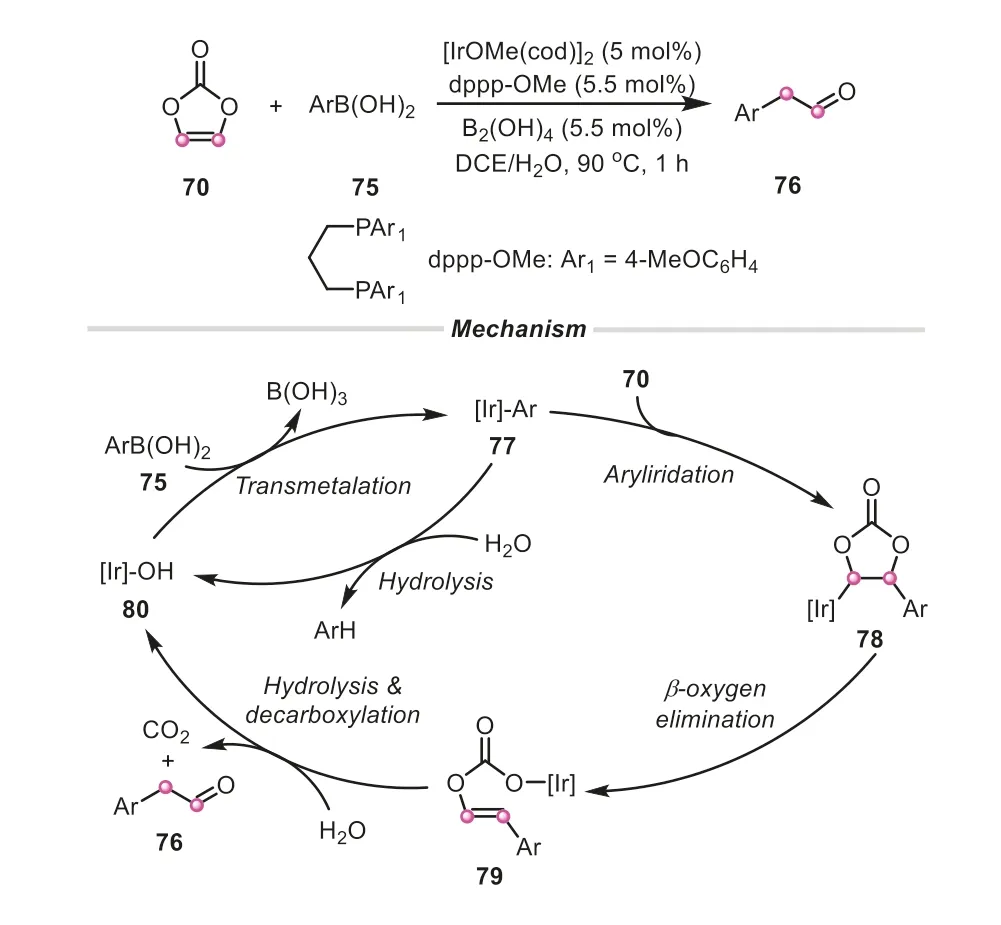
Scheme 19.Vinylene carbonate as ethynol equivalent for the synthesis of arylacetaldehydes.

Scheme 20.Vinylene carbonate as ethynol equivalent for the synthesis of fused quinazolinones.
Since 2019,Nishii and Miura group disclosed an intriguing discovery in vinylene carbonate based on C-H activation,where vinylene carbonate can behave as a “vinylene transfer” agent[36,42,43].This group made a report on Rh-catalyzed C-H/N-H annulation with vinylene carbonate as an acetylene equivalent for the synthesis of variousN-heteroaromatics (Scheme 21) [36].The prominent feature of this protocol was to circumvent the addition of external oxidant or base.It was proposed that the amide directing group initially coordinated with a cationic Cp*Rh(III) species,giving a five-membered rhodacycle85.And the subsequent migratory insertion of vinylene carbonate provided a seven-membered metallacycle86,which was accompanied by a consecutive reductive elimination of C-N and oxidative addition of Cp*Rh to the adjacent C-O bond,affording the complex88.Finally,the occurrence ofβ-oxygen elimination of88could liberate the coupling product.Following the same principle,similar C-H/C-H,and C-H/OH oxidative annulation strategies using vinylene carbonate as an acetylene surrogate were further developed to produce imidazolefused aromatics and isocoumarin products by the same group(Scheme 22) [42,43].
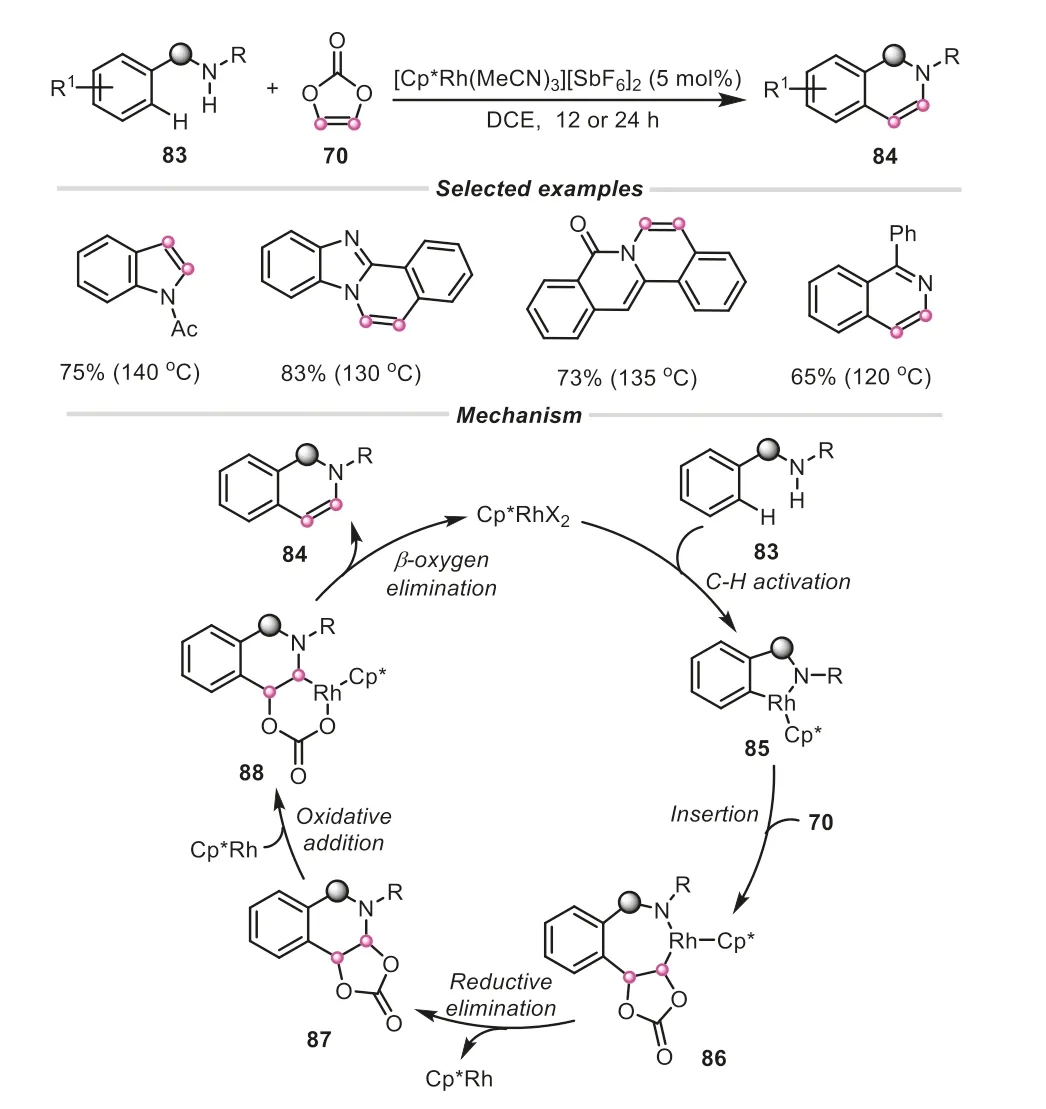
Scheme 21.Vinylene carbonate as acetylene equivalent for the synthesis of various N-heteroaromatics.
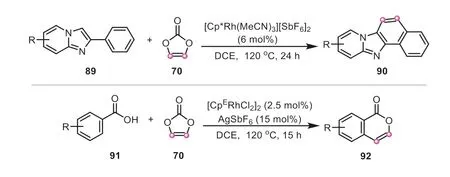
Scheme 22.Vinylene carbonate as vinylene transfer agent for the annulation of imidazopyridines and isocoumarins.
Generally,most of the above-mentioned transformations adopting vinylene carbonate as the acetylene equivalent was based on the use of precious Rh-based catalysts.To optimize and expand the catalytic system,in subsequent research,Xiao and Zhang demonstrated a cost-effective Co(III)-catalyzed C-H activation/annulative vinylene transfer strategy in 2,2,2-trifluoroethanol (TFE) for the synthesis of isoquinolinones and pyridinones (Scheme 23) [37].Similar to Rh-catalyzed mechanism,amides firstly underwent CH activation with the assistance of cationic cobalt species,followed by a sequential process,including the insertion of vinylene carbonate,the reductive elimination and oxidative addition of Cp*Co,andβ-oxygen elimination to generate target products.

Scheme 23.Co(III)-catalyzed annulation for the synthesis of isoquinolinones and pyridinones with vinylene carbonate as acetylene surrogate.
Most recently,the metal-free catalytic system has been developed for vinylene carbonate-involved transformations.In 2022,Cao and Liu group developed a direct and effective approach based on the [3+2] annulation of azomethine imines and vinylene carbonate for the divergent synthesis of pyrazolo[1,5-a]pyridine framework [44],which is an important prevalent structure and has been displayed in numerous pharmaceuticals and biologically active molecules (Scheme 24).WhenN-aminoazinium salts were used as the coupling partner,the reaction proceeded smoothly without any catalyst and additive,in which vinylene carbonate served as the role of acetylene by the release of carbonate anion to furnish pyrazolo[1,5-a]quinolines.On the contrary,Niminoquinolinium ylides reacted with vinylene carbonate to generate pyrazolo[1,5-a]quinolin-3-ol compounds,in which vinylene carbonate acted as an ethynol surrogateviadecarboxylation in the presence of K2CO3.In addition,the reaction of isoquinolinium salt with vinylene carbonate also proceeded effectively,affording pyrazolo[5,1-a]isoquinolin-1-ol in satisfactory yields.These results revealed that the counterparts gave a significant influence on the functions of vinylene carbonate performed in the transformations.

Scheme 24.Metal-free-catalyzed [3+2] cyclization of azomethine imines with vinylene carbonate as acetylene or ethynol surrogate.
In the aforementioned works,vinylene carbonate worked well as an “ethynol” or “acetylene” unit.Apart from these,vinylene carbonate can be also controlled as an efficient glyoxal equivalent.For example,Kimetal.developed an expedient palladium-catalyzed method for the synthesis of benzils from aryl bromides and vinylene carbonate [45].As shown in Scheme 25,the reaction might be initiated by a syn-carbopalladation of the PhPdBr complex to vinylene carbonate,which was followed by an elimination of Pd(0) to produce the intermediate103.Afterwards,103would go through a facile deprotonation to obtain monoaryl substituted vinylene carbonate104.Then a similar arylation occurred once more,generating105.In the assistance of base,105underwent a ring-opening to form theα-hydroxyl ketone106,which was subsequently transformed into the desired benzil productviaan aerobic oxidation.Compared to commonly used oxidation of alkynes,the present protocol provided a more simple and useful methodology for the preparation of benzil derivatives.
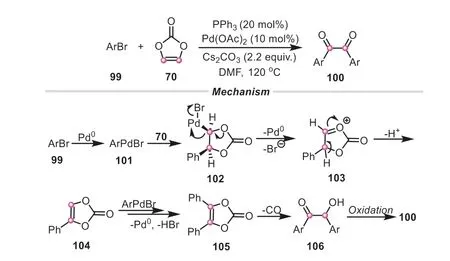
Scheme 25.Vinylene carbonate as glyoxal equivalent for the synthesis of benzils.
With the intensive research on vinylene carbonate,it has been proposed that vinylene carbonate can serve as an acylating reagent to accomplish some transformations.For example,in 2021,Qian and Cheng developed a facile rhodium-catalyzed annulation from amidine with vinylene carbonate,in which vinylene carbonate could provide an acetyl fragment to promote the construction of quinazoline ring (Scheme 26) [46].The kinetic experiments inferred that the cleavage of theortho-C-H bond might be the ratedetermining step of the reaction.Based on this,a possible reaction process was proposed as follows: an initial coordination of Rh and amidine produced a metal complex109,which was accompanied by a migratory insertion of vinylene carbonate to afford the rhodacycle110.After that,110was converted into the intermediate112through a sequentialβ-H elimination,insertion of HRhCp*andβ-O elimination.Then112underwent a rapid protonolysis to result in the enol intermediate113,along with the release of catalyst and CO2.Subsequently,a tautomerization proceeded to render an acetophenone-type intermediate113′,which easily went through an intramolecular cyclization to get the final 4-methylquinazoline product.
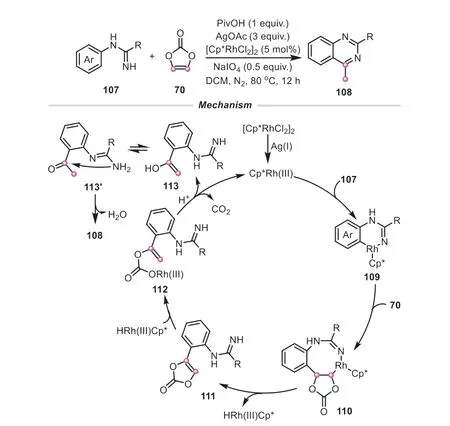
Scheme 26.Vinylene carbonate as an acylation reagent for the synthesis of 4-methylquinazolines.
In the same year,Nan and Ma group reported a formal [5+1]cyclization towards the assembly of various 4-methylpyrrolo[1,2-a]quinoxalines by the combination of vinylene carbonate with pyrrolyl/indolylanilines (Scheme 27) [47].In a similar manner,vinylene carbonate served well as an elegant “acetyl” substitute,giving a C1 cyclic unit to participate in the transformation.Notably,this reaction was conducted in a quite simple reaction system,avoiding the use of external oxidants,additives and bases.In addition,this protocol also performed good functional group tolerance and excellent yields,rendering it more practical.
Scheme 27.Vinylene carbonate as an acylation reagent for the synthesis of 4-methylpyrrolo[1,2-a]quinoxalines.
With the development of multiple reactivities of vinylene carbonate,the assembly of diverse reactivities into the same molecule has become an attractive topic for the organic community.In 2021,vinylene carbonate acting as a difunctional coupling partner was firstly reported by Nanetal.for the preparation of 2-methylquinoline derivatives (Scheme 28) [48].In the tandem cyclization,two molecules of vinylene carbonate were successively combined with anilines,in which vinylene carbonate served as a dual synthon,providing two different C2 units “vinylene” and“acetyl” concurrently.The kinetic experiment suggested that the CH cleavage might not be involved in the rate-determining step owing to a low KIE value.Besides,the addition of D2O led to a significant deuteration incorporation,which indicated that a reversible metalation process might be involved in the process.Notably,the electron-rich anilines outperformed electron-deficient ones in this transformation,which also supported that the arylrhodium species was produced probably by direct electronic metalation.
Scheme 28.Vinylene carbonate as a dual C2 synthon for the synthesis of 2-methylquinolines.
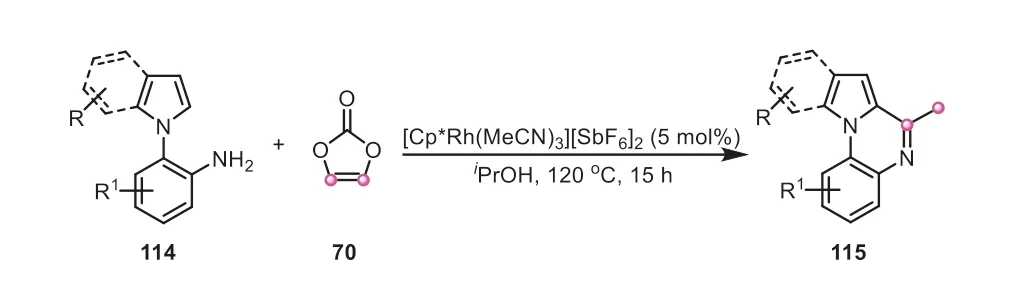
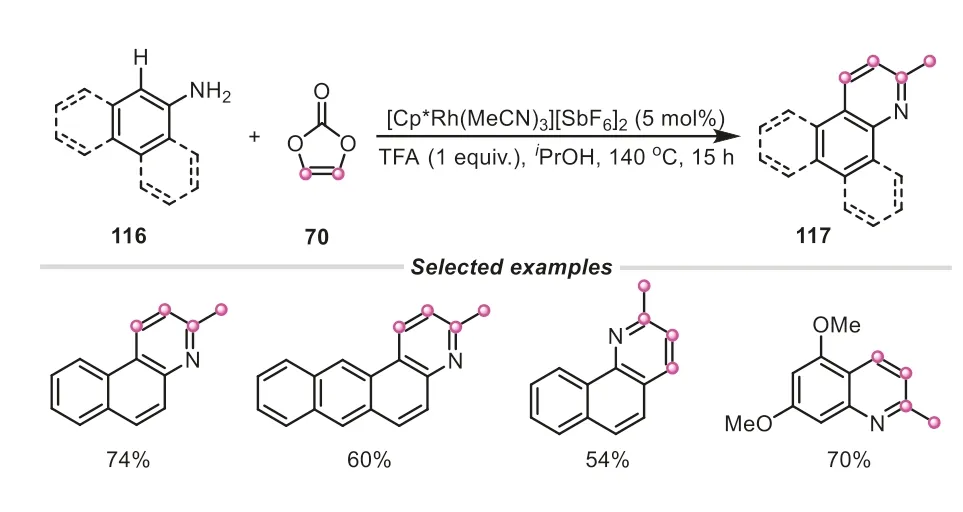
Later on,vinylene carbonate has been also used as both C1 and C2 dual synthons by Pi and Cui group for the construction of complex four rings-fused indazolo[2,3-a]quinolines,in which three C-C bonds and C-N bond were formed (Scheme 29) [49].As demonstrated in the proposed mechanism,vinylene carbonate played a dual role,acting as both an acylation reagent and “vinylene transfer” agent in the reaction.The vinylene carbonate initially united with azobenzene,giving the acetyl-substituted azobenzene122,which could be detected by HRMS.Intriguingly,the compound122underwent an oxidative C-C cleavage,and intramolecular nucleophilic cyclization to afford 2-phenyl-2H-indazole123,in which vinylene carbonate actually provided a carbon atom to incorporate into the skeleton of the intermediate123.Afterwards,another molecule of vinylene carbonate offering vinylene fragment was combined with123,successfully generating the desired indazolo[2,3-a]quinoline product.All these examples demonstrated that vinylene carbonate is an elegant versatile reagent for creating and enriching the structural complexity in organic synthesis.
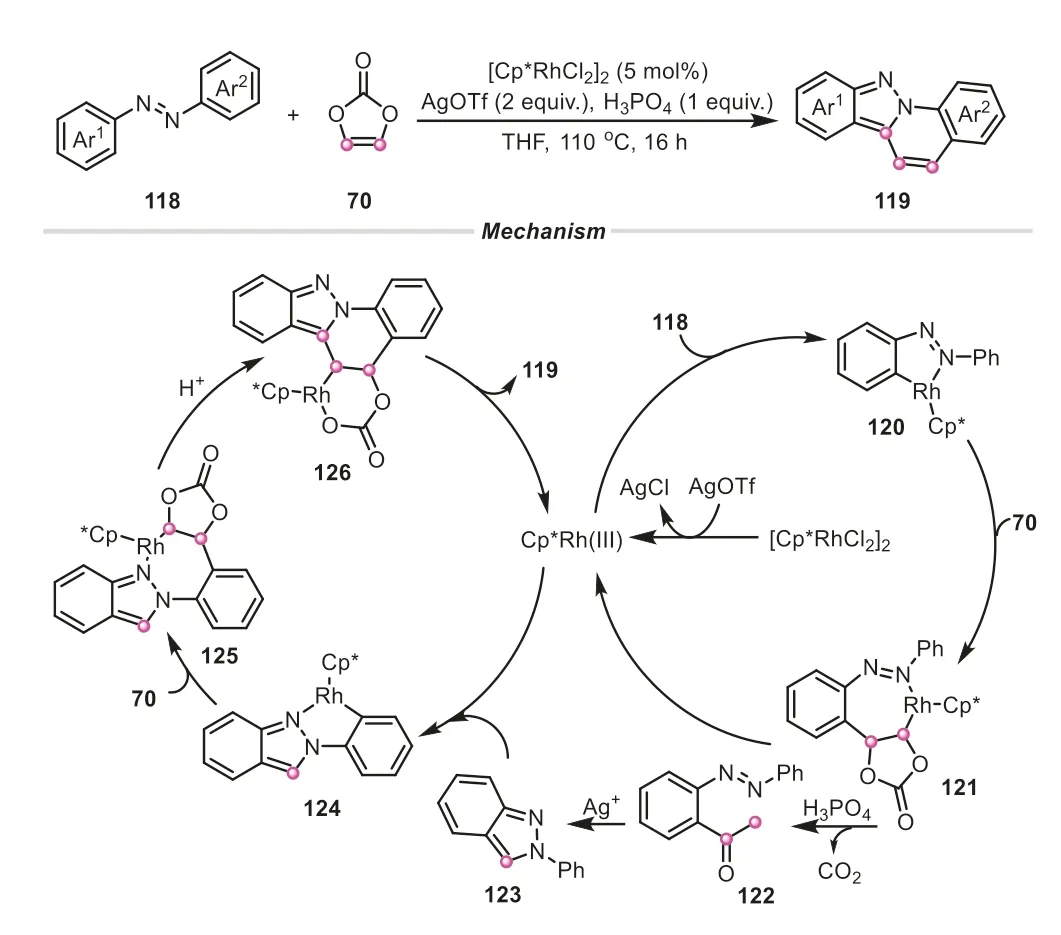
Scheme 29.Vinylene carbonate as a dual C1/C2 synthon for the synthesis of indazolo[2,3-a]quinolines.
5.Vinyl azides
Vinyl azides are a category of commercially available and energetic reagents,and they can exhibit distinct and unprecedented reactivity towards the synthesis of a great many of nitrogencontaining heterocycles [50].Owing to the existence of the azide group tethered to an alkene moiety,vinyl azides can play many different roles under different reaction conditions,such as being a nucleophile,an electrophile,or a radical acceptor.Vinyl azides can undergo various bond cleavage modes,such as N-N,C-N,C=C,and C-C,which rendered vinyl azides to serve as multipurpose precursors for providing various units (Scheme 30).Therefore,there is no doubt that vinyl azides have become a significant class of multifunctionalized reagents in organic synthesis.

Scheme 30.Vinyl azides as versatile synthons via multiple bond scissions.
Among those fragments,it is very common that vinyl azides act as a pivotal three-atom synthon for the construction of various azaheterocycles through the cleavage of N-N bond with the release of N2.For example,Jiaoetal.developed a novel and efficient disubstituted pyrroles synthesis through the denitrogenative annulation ofα-azidostyrene with aryl acetaldehydes (Scheme 31) [51].With this methodology,the divergent substituted pyrroles were obtained with satisfactory yields and high regioselectivities by simply switching transition-metal catalysts.The use of copper acetate as the catalyst directly led to the formation of 2,4-disubsituted pyrroles in the mixed solvent DMSO/EtOH,while nickel chloride selectively facilitated the generation of 3,4-diaryl pyrroles inN,Ndimethylacetamide (DMAc).For the formation of 2,4-disubsituted pyrrole (path a),it was proposed that CuI species was initially generated by the reduction of DMSO/EtOH,which then led to the formation of the radical intermediates130through the denitrogenative decomposition of vinyl azide.The subsequent radical coupling between130and the enol tautomer from the phenylacetaldehydes proceeded to furnish the intermediates132,which underwent an intramolecular nucleophilic cyclization,delivering the intermediate133.Afterwards,the successive protonation/dehydration afforded the target 2,4-disubsituted pyrrole.Different from the coppercatalyzed denitrogenative decomposition of127,nickel catalyst initially promoted the generation of the strained three-membered 2H-azirines intermediate135(path b),an equivalent of vinyl nitrenes.Subsequent nucleophilic attack from134and ring-opening reaction produced a five-membered species137,which was followed by aβ-OH elimination and tautomerization,yielding 3,4-disubstituted pyrrole.Even thoughα-azidostyrene provided the three-atom fragments in both of the two transformations,the intermediatesinsituinduced by different reaction conditions led to the formation of distinct subunits,successfully accomplishing the synthesis of pyrrole with different substituent patterns.

Scheme 31.Vinyl azides as three-atom synthons for the synthesis of disubstituted pyrroles.
A similar strategy was also applied for the divergent synthesis of 2-aminothiazole derivatives from vinyl azides and potassium thiocyanate by Zhang and Yu group,in which the type of 2-aminothiazole products could be effectively controlled by the choice of catalysts (Scheme 32) [52].In spite of two different pathways involved in these transformations,the vinyl azides played the same role,both providing C-N-C-type fragment.For the Pd(OAc)2-catalyzed reaction (path a),an ionic pathway proceeded to give the 4-substituted 2-aminothiazoles.The coordination of palladium(II) to vinyl azide generated an electrophilic species142.The subsequent nucleophilic attack of thiocyanate anion promoted the scission of N-N,producing the intermediate143with the release of N2.After that,a consecutive intramolecular cyclization/protonation/tautomerization occurred to afford 4-substituted 2-aminothiazoles.In the FeBr3-promoted reaction (path b),a radical mechanism was proposed.Initially,the single-electron transfer (SET) between thiocyanate anion with Fe(III) resulted in Fe(II) and thiocyanate radical.Fe(II) would assist the conversion of vinyl azide into iminyl iron(III) radical145viathe extrusion of N2,which was combined with thiocyanate radical,delivering the intermediate146.Subsequently,the intermediate146underwent nucleophilic cyclization and 1,5-Hshift to obtain the 2-aminothiazole intermediate148.It was worth mentioning that the thiocyanate radical would attack the electron-rich site of148to generate the radical species149,which could be readily transformed into the cation150in the presence of Fe(III) and O2.At last,the deprotonationinduced aromatization proceeded to form the desired 4-substituted 5-thiocyano-2-aminothiazole141.Apparently,the simple starting materials,potassium thiocyanate,also performed its dual functionalization under the mild conditions,not only acting as a two-atom fragment converging into the thiazole skeleton,but also offering a thiocyano,which made this protocol quite useful and attractive.Furthermore,vinyl azides serving as the three-atom synthon can be used for the preparation of 2H-imidazoles and pyridines [53,54].
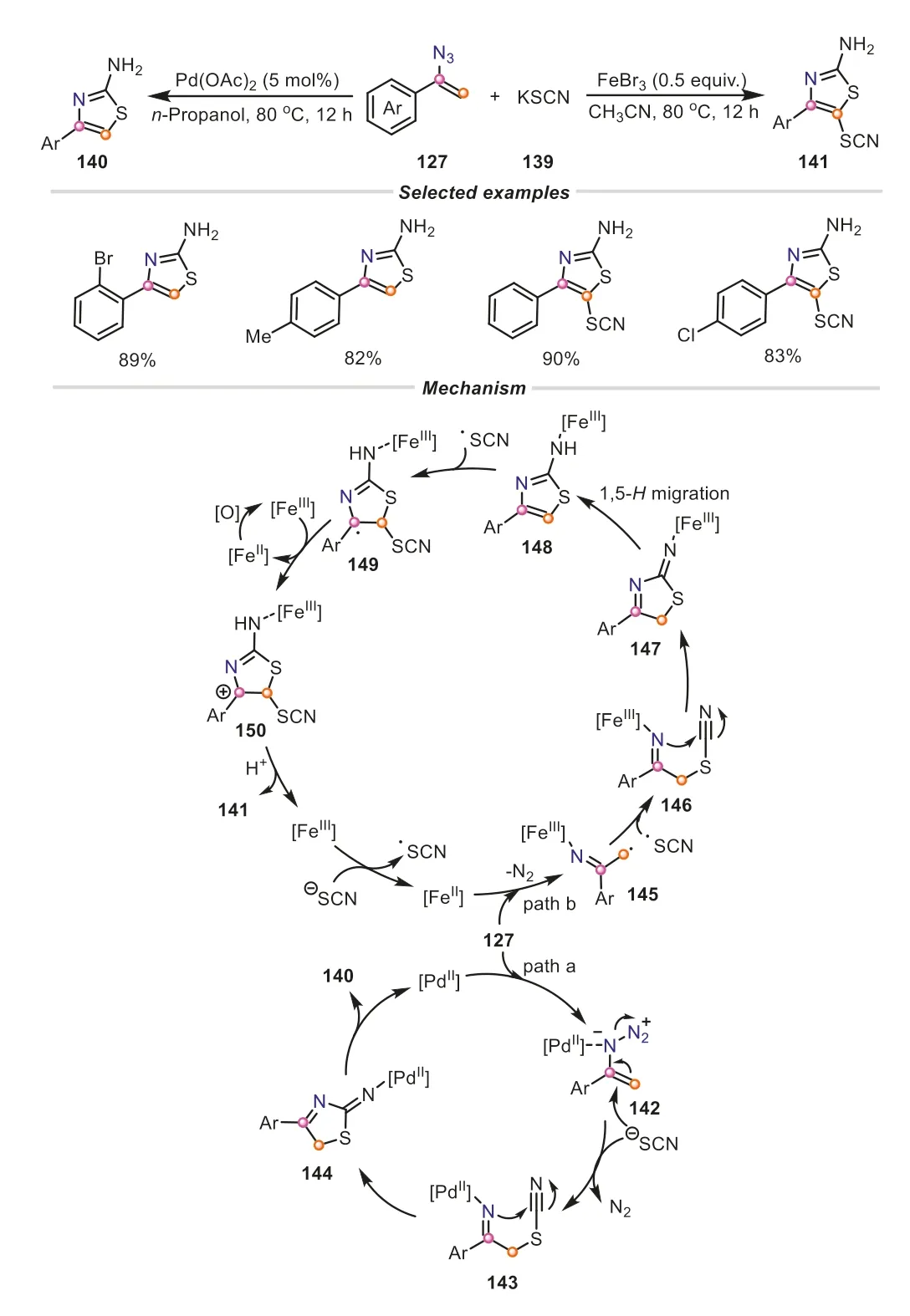
Scheme 32.Vinyl azides as three-atom synthons for the divergent synthesis of 2-aminothiazole derivatives.
Vinyl azides can be regarded as enamine equivalents to perform their potential nucleophilicity.In 2014,Chiba and co-workers reported a BF3·OEt2-mediated amides synthesis from vinyl azides and various carbon-based electrophiles (Scheme 33) [55].In these reactions,the initial nucleophilic attack of vinyl azides towards electrophiles produced iminodiazonium ions157,which subsequently underwent Schmidt-type 1,2-migration to give nitrilium ions158.The final hydrolysis of158delivered the corresponding secondary amides.During this process,α-aryl vinyl azides actually went through a successive N-N bond and C-C bond cleavages to provide nitrilium ions,which ensured the final formation of amides.Intriguingly,whenα-alkyl-substituted vinyl azide reacted with imine151,a mixture of amide and dihydroimidazole was obtained in a combined yield of 68% (ratio=1/1).This result indicated that two isomers (E-andZ-isomers) of iminodiazonium ions might be formed,after which 1,2-migration would occur on two different substituents,consequently affording two constitutional isomers nitrilium ions.Of note,the ratio of final products might be influenced by the migratory aptitude of substituents and the reaction conditions to a large extent.

Scheme 33.Vinyl azides as enamine-type nucleophiles for the synthesis of amides.
Prompted by the above work,the same group employed the electrophiles tethered with a nucleophilic site to react with vinyl azides,in which the resulting iminodiazonium ions could be trapped by the nucleophilic reactive site,prior to the substituent migration,generating cyclic structures [56].The reaction of vinyl azides withN-phenyl aldimines delivered a variety of quinolines with good to excellent yields (Scheme 34).When theN-alkenyl aldimines were used as annulation partners,the corresponding tetrasubstituted pyridines were obtained in the presence of DDQ as oxidant.It should be mentioned that whether aryl or alkyl substituted vinyl azides,even heteroaryl vinyl azides,were also compatible with this protocol,furnishing the corresponding products with good selectivity.In these transformations,vinyl azides accomplished the supply of vinyl fragmentsviathe cleavage of C-N bond.
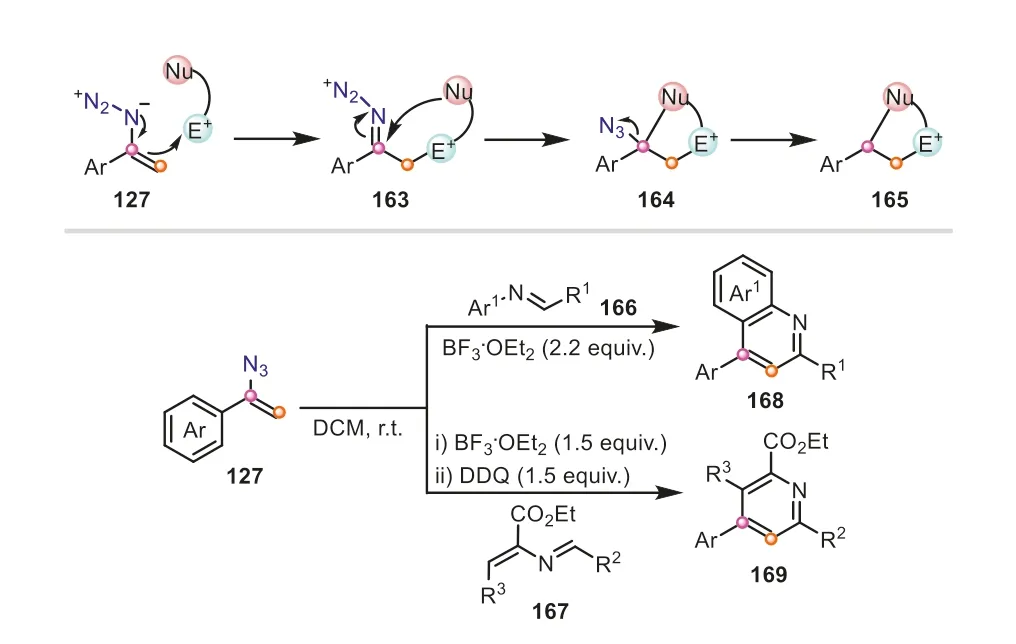
Scheme 34.Vinyl azides as vinyl surrogates for the synthesis of quinolines and pyridines via [4+2] annulation.
In addition,vinyl azides were also proven to be an effective C1-methylene donor through the cleavage of C=C bond.In 2015,Liu’s group unprecedentedly developed a gold-mediated C=C cleavage of vinyl azides for the synthesis of buta-1,3-dien-2-yl esters by adding the methylene to the terminal carbons of alkynyl in propargyl esters (Scheme 35) [57].For monoaryl-substituted propargylic esters,Z-configured buta-1,3-dien-2-yl esters were selectively formed,in which the gold-assisted 1,2-carboxylate shift might proceed to form the carbene174.Notably,the hydrogen located in174cisto gold fragments would minimize the steric hindrance.The subsequent attack of C=C from vinyl azides on the intermediate174was expected to generate intermediate175.After that,175released a molecule of N2,which further induced the single CH2-CPh bond cleavage to deliver theZ-configured buta-1,3-dien-2-yl ester product171,with benzonitrile172as the byproduct.However,it was observed thatE-configured products were exclusively generated for alkynyl-and alkenyl-substituted propargyl esters,which might be explained by a remote interaction resulting from the existence ofπ-bond motifcisto gold in the initial gold-carbenes.Furthermore,3,3-disubstituted propargyl esters were also favorable for the formation ofE-configured products.

Scheme 35.Vinyl azides as methylene donors for the synthesis of buta-1,3-dien-2-yl esters.
Based on the aforementioned results,Katukojvalaetal.envisaged that the combination of the C1-methylene and the threeatom N-C-C-fragment provided by vinyl azides could be used for the construction of other novel azaheterocycles.In 2018,this group disclosed a novel and concise dirhodium-catalyzed [1+1+3]annulation of vinyl azides and diazoenals,which successfully rendered the synthesis of valuable enal-functionalized 1-pyrrolines(Scheme 36) [58].The reaction could be efficiently achieved by simple operation at room temperature with the assistance of 3 mol% of dirhodium carboxylate.It was proposed that the reaction was initiated with the Rh(II)-catalyzed denitrogenation of diazo compound,giving an electrophilic rhodium carbenoid178,which was followed by a nucleophilic addition of vinyl azide to form a crucial acyclic zwitterion179.Triggered by the departure of N2,179underwent a C-C bond fragmentation under the assistance of rhodium catalyst,delivering olefin180and nitrile172.Afterwards,the [2+2]-cycloaddition of180with another molecule of127occurred to produce the strained cyclobutane181.The subsequent loss of nitrogen led to the ring expansion of181by the migration of the quaternary carbon,furnishing the target product 1-pyrroline.As expected,two molecules of vinyl azides were involved in the transformation,one promoting the methylenation of diazo compoundsviathe C-C cleavage,and another one generating three-atom N-C-C-fragment to couple with theinsituformed olefin through the N-N fragmentation.
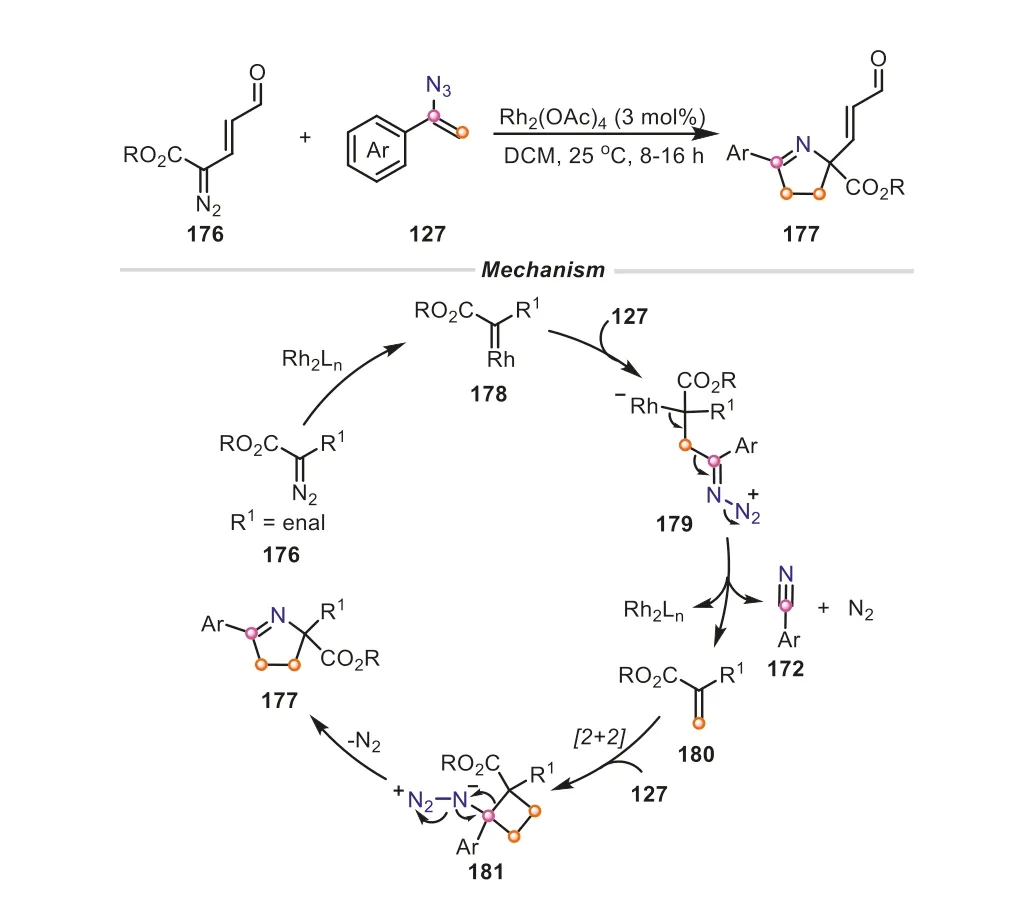
Scheme 36.Vinyl azides as dual methylene and three-atom synthons for the synthesis of 1-pyrrolines.
In the same year,viny azides acting as the dual surrogates of methylene and styryl were also reported by Yang and Jiang group to assemble 4-substituted quinolines (Scheme 37) [59].Two types of cleavage modes of vinyl azides were present in the transformation.As illustrated in Scheme 37,the electrophilic C=C bond of viny azide was attacked by aniline in the presence of zinc catalyst,followed by the loss of nitrogen,generating the intermediate185.In a similar manner,the intermediate185was converted to the imine186through the C-C bond scission,generating benzonitrile as a by-product.Subsequently,the [4+2] annulation of186and viny azide gave the intermediate189with the elimination of HN3,which further underwent an aromatization,eventually delivering the desired quinoline product.More interestingly,the crossover reactions of two different vinyl azides with anilines also proceeded smoothly,selectively yielding 2,4-disubsituted quinolines as the main products.This reaction featured distinct advantages,such as simple operation,mild reaction conditions,and high step economy,as well as the use of environmentally friendly air as oxidant.Undoubtedly,these research works opened up a new window for the construction of nitrogen-containing heterocycles by employing vinyl azides as simple starting material.

Scheme 37.Vinyl azides as vinyl surrogates for the synthesis of quinolines.
6.Aryldiazonium salts
Aryldiazonium salts (ArN2X) are a class of attractive chemicals because of their ready accessibility and versatile reactivities,which have been recognized as useful precursors spread in a large number of organic transformations [60,61].Aryldiazonium salts can be generally obtained from low-cost and abundantly available anilines and nitrites with high yields.Their stability is highly associated with the counterion.It has been confirmed that the less oxidable and non-nucleophilic anions,such as tetrafluoroborate (BF4-),hexafluorophosphate (PF6-) and tosylate (OTf-),can reveal good aptitude in stabilizing arenediazonium cations [62].And the stable crystalline salt,ArN2BF4,is the most frequently employed one so far.Owing to the strong tendency to remove N2,diazonium salts have been engaged as aryl precursors for the substitutiontype functionalizations (Scheme 38) [63].In addition,aryldiazonium salts can serve as highly reactive electrophiles or radical receptors for various addition reactions with the retention of N-N moiety [64].Most importantly,the use of aryldiazonium salts as dual synthons has emerged successively.For these reasons,aryldiazonium salts have been considered as a category of versatile reagents for organic transformations.

Scheme 38.Aryldiazonium salts as aryl precursors via the cleavage of C-N bond.
As the commonly used aryl radical precursors,aryl diazonium salts have been widely applied in constructing the aryl C-C bonds.As exemplified in Scheme 39,aryldiazonium tetrafluoroborates can go through radical addition with different double bonds,C=C and C=N,affording a variety of arylated molecules.And the aryl radical intermediates can beinsitugenerated by multifarious catalytic routes,including transition-metal catalysis,photocatalysis,and electrocatalysis.The Pd(OAc)2-mediated Heck-Matsuda reaction of ethenesulfonyl fluoride191and aryl diazonium tetrafluoroborates smoothly furnishedβ-arylethenesulfonyl fluorides192under mild reaction conditions [65],which could be used as versatile biselectrophiles for the click chemistry of sulfur(VI) fluoride exchange.In addition,the ferrocene-promoted regioselective arylation can lead to the modification of BODIPY dyes (4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes),allowing for the preparation of brightly fluorescent arylated boron dipyrrins194[66].In the lightinduced protocol for the synthesis of stilbenes,the blue LED can be directly used as the light source with the assistance of Eosin Y as the photosensitizer [67].For the electrochemical cross-coupling of quinoxalin-2(1H)-ones,the transformations could be achieved by carrying out a constant current electrolysis of 4 mA/cm2in a simple undivided cell without external electrolyte [68].These examples elegantly performed the effectiveness and diversity of catalytic systems in the aryldiazonium salts-initiated radical arylations.
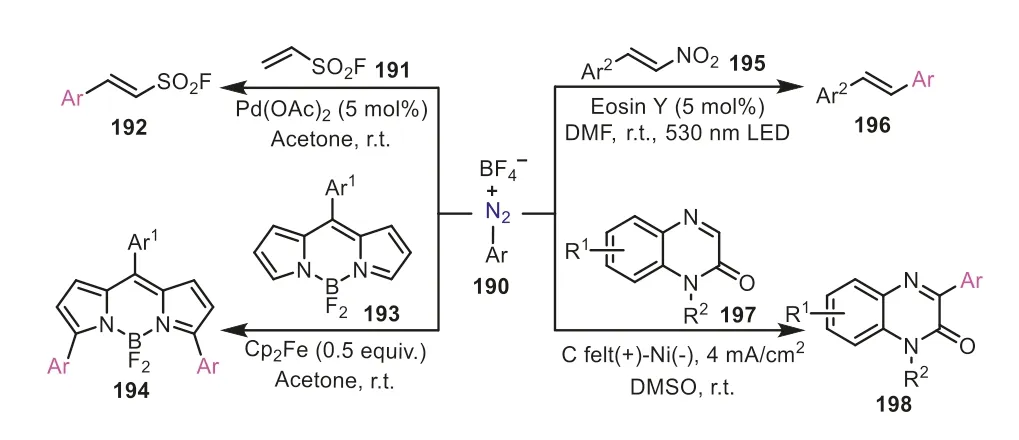
Scheme 39.Aryldiazonium salts as aryl donors for the construction of C-C bonds.
Most recently,a transition metal-free dioxygenative reaction between aryldiazonium salts and aryl alkynes for the assembly of vicinal diketone was developed by Cheng and Wan (Scheme 40)[69].Interestingly,the introduction of 4 ˚A MS and the use of the mixture of MeCN and DMSO as solvent could improve the yield of diketones under mild room temperature conditions.In the Eosin Y-based photocatalysis,Eosin Y was initially excited to Eosin Y*.The combination of Eosin Y*with diazonium salt generated an aryl radical by releasing nitrogen,which was followed by an addition to alkyne,providing intermediate201.The generated201was subsequently coupled with molecular oxygen to give rise to a peroxide cycle203involving a peroxide radical202.The final ring opening of203afforded the desired diketone product.This synthetic strategy provided a simple and green approach to prepare diverse 1,2-diaryl diketones with both symmetrical and unsymmetrical structures by employing clean molecular oxygen as the O-atom source.

Scheme 40.Aryldiazonium salts as aryl donors for the synthesis of vicinal diketones.
Apart from the synthesis of linear molecules,the aryl radicalsinduced cascade reactions can be used for the construction of cyclic compounds.In 2022,Singh and co-workers achieved the synthesis of stereospecifictrans-oxiranes from cinnamic acids and aryldiazonium salts through the decarboxylative epoxidation process (Scheme 41) [70].The reaction proceeded under the cocatalysis of visible-light and CuCl with TBHP as the oxygen source.Also,the transformation was featured with good compatibility towards diverse functional groups and a wide range of substrates were well tolerated under the reaction conditions.As described in Scheme 41,the aryldiazonium salt was converted to the aryl radical by SET with the aid of Eosin Y.Subsequently,the reaction of the aryl radical with the cinnamate gave the intermediate207,which delivered a five-membered chelate ring208under the catalysis of Cu(II) generated by the SET from Cu(I) and Eosin Y+·.Afterwards,208was converted to 1,2-dioxolan-3-one210passing through an intermediate209in the presence of DBU and TBHP.Eventually,1,2-dioxolan-3-one decarboxylate produced the desired product205.In a similar mechanism,aryl radicals-triggered cyclizations were reported for the synthesis of 3,3-disubstituted indolinones,isochromanones and isoflavones (Scheme 42) [71-73].
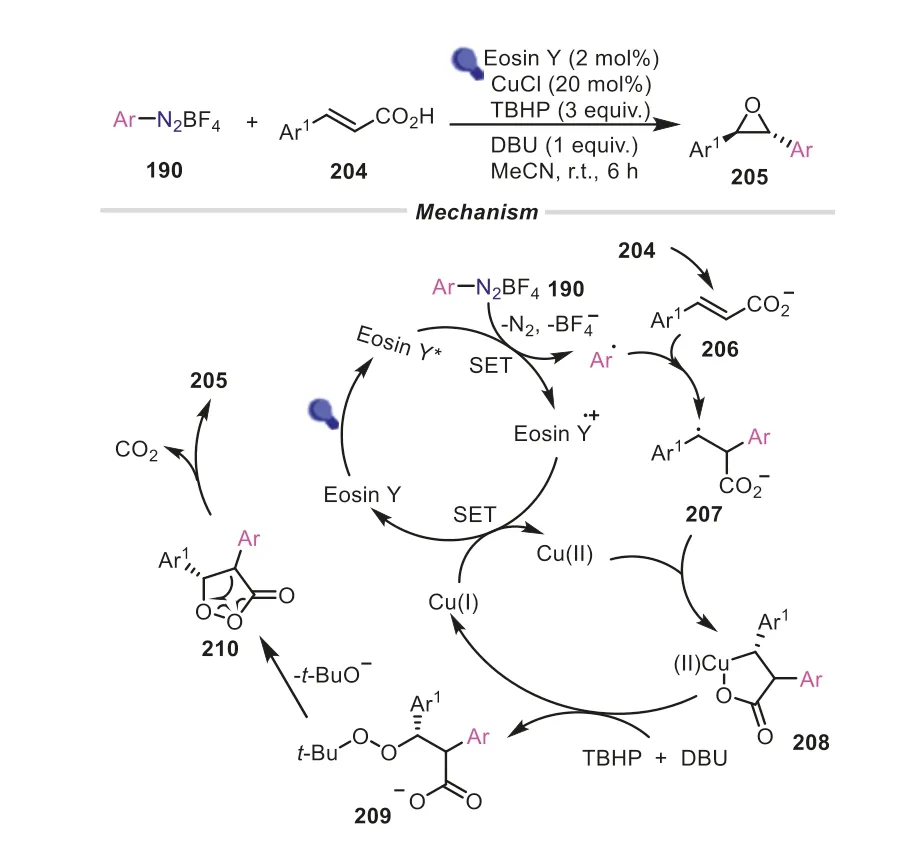
Scheme 41.Aryldiazonium salts as aryl donors for the synthesis of trans-oxiranes.
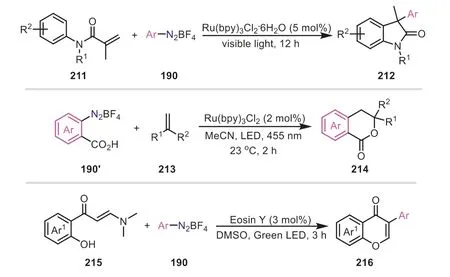
Scheme 42.Aryl radicals-induced cascade reactions for the construction of cyclic compounds.
Additionally,arene diazonium salts have also enjoyed numerous applications with the retention of the nitrogen group.The aryldiazonium cation can be easily intercepted by various nucleophiles,such as enolate species,isocyanides,diazo compounds,enamines,thus generating diverse linear or cyclicN-containing compounds (Scheme 43).In 2018,Zhu and Jiang reported a basecatalyzed intermolecular C(sp3)-H amination reaction of aryl diazonium salts and trifluoromethyl ketones under mild reaction conditions,obtaining trifluoroacetylated hydrazones218in excellent yields with excellentE/Zselectivity [74].In the same year,a catalyst-dependent regioselective cycloaddition of aryldiazonium salts and isocyanides was applied for the facile construction of functionalized 1,2,4-triazoles [75].This reaction smoothly produced 1,3-disubstituted 1,2,4-triazoles221under the catalysis of Ag(I),whereas the Cu(II) catalysis led to 1,5-disubstituted 1,2,4-triazoles220with moderate to good yields.Similarly,aryldiazonium tetrafluoroborate could be also combined with trifluorodiazoethane to generate 2,5-disubstituted tetrazoles223[76].Moreover,the functionalized tetrazoles225can be also achieved by employing amidines as the nitrogen-based nucleophiles to trap aryldiazonium cation,which has been reported by Liu and co-workers in 2015 [77].The sequential reaction of aryldiazonium salts with amidines was performed in one-pot two-step manner under the assistance of K2CO3,followed by the treatment of I2/KI to afford several 2,5-disubstituted tetrazole products.In these transformations,the key step was the addition of nucleophiles to diazonium cations,rendering diazonium salts to serve as the resource of two N atoms to offer diversiform azole products.
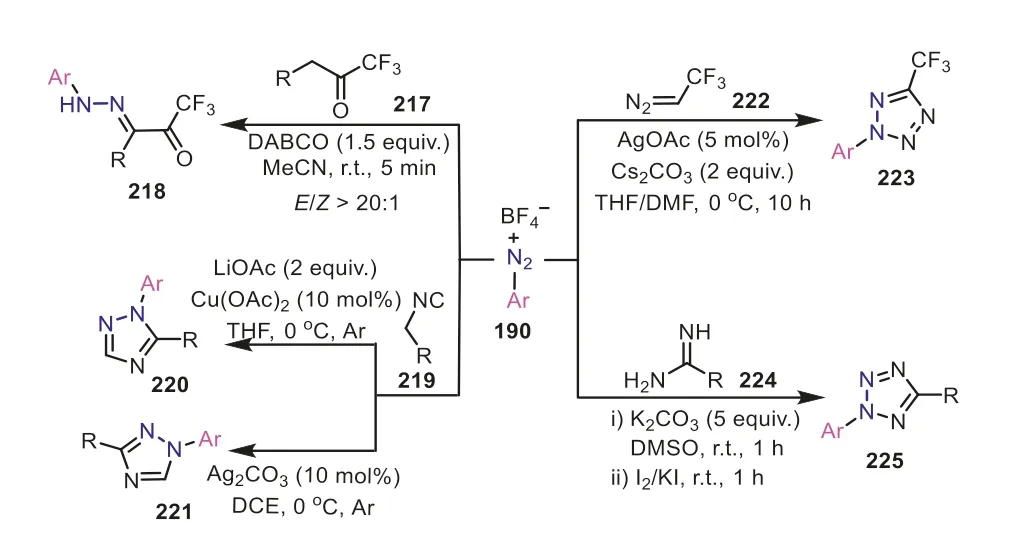
Scheme 43.Aryldiazonium salts as nitrogen sources for the synthesis of Ncontaining compounds.
TheN-containing coupling reactions can be also obtained through the radical-type interception of arene diazonium ions,in which theinsitugenerated diazonium radical cations were accompanied by a sequential SET or radical-chain process to furnish final products.In 2014,Antonchicketal.devised a novel radical-induced multicomponent cascade reaction of diazonium salts,alkenes,and sodium triflinate,providing a straightforward route to access a wide range of trifluoromethylated indoles (Scheme 44) [78].The protocol was initiated by the formation of an electrophilic trifluoromethyl radical,followed by an addition to alkene,producing radical intermediate229.Subsequently,the radical229was readily captured by aryldiazonium salt to give radical cation230,which underwent a successive SET and [1,3]-hydride shift to provide an arylhydrazone intermediate231′,which was a crucial intermediate during the classical Fischer indole synthesis process.Subsequently,231′was transformed into the desired trifluoromethylated indole product through a successive [3,3]-sigmatropic rearrangement/cyclization/ammonia elimination process.Different from the above radical mechanism,Knochel and co-workers employed functionalized alkylzinc reagents as the counterpart of aryldiazonium salts to undergo a sequential Japp-Klingemann reaction/isomerization/Fischer cyclization,offering a novel alternative to polysubstituted indoles (Scheme 45) [79].Owing to the occurrence of ammonia elimination in the two transformations,aryldiazonium salts acted as the arylamine surrogate to incorporate into the final indole scaffolds with good chemoselectivity.
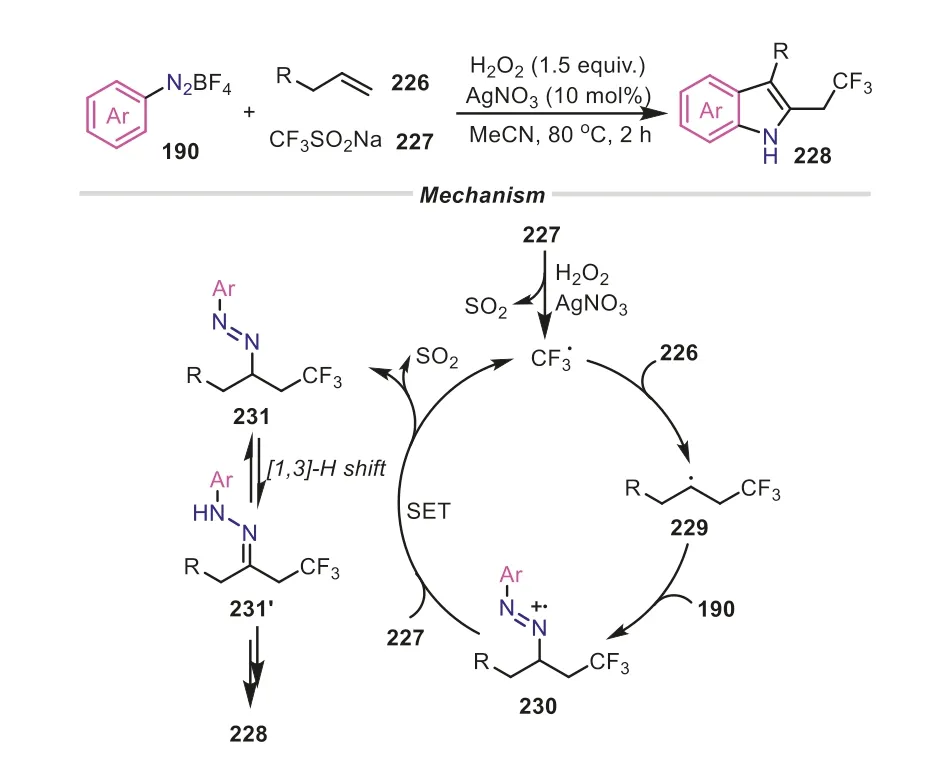
Scheme 44.Aryldiazonium salts as arylamine surrogates for the synthesis of indoles through a radical pathway.
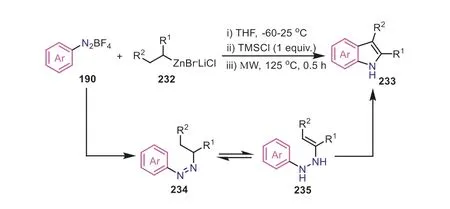
Scheme 45.Aryldiazonium salts as arylamine surrogates for the synthesis of indoles through an ionic pathway.
In 2017,Heinrich’s group developed a transition-metal-free Meerwein-type carboamination of inert alkenes with aryldiazonium tetrafluoroborate salts,enabling the formation of azo compounds in moderate to good yields (Scheme 46) [80].The reaction initially proceeded through the slow generation of aryl radicals,followed by a radical addition to form an alkyl radical.The subsequent trapping of alkyl radical by another molecule of aryl diazonium ion was a crucial step,which then went through SET to afford the difunctionalization products.In the subsequent research,frozen aryldiazonium chlorides have been also successfully applied in the radical alkene functionalization,resulting in azo products with moderate yields.
In 2019,Wu’s group reported a rongalite-mediated radical annulation protocol for the selective assembly of polysubstituted pyrazoles fromα,β-unsaturated carbonyls and aryl diazonium salts at mild room temperature (Scheme 47) [81].During this protocol,aryl diazonium salts were engaged as the dual precursors of both aryl and arylhydrazine to participate in the cascade cyclization.In a similar manner,the aryl radical was firstly formed with the aid of rongalite,followed by a Meerwein-type arylation to provide benzylic radical240.The radical240was easily trapped by aryl diazonium cation to go through a SET and reduction,affording the hydrazine intermediate243.The final annulation and aromatization produced the target pyrazole product.

Scheme 47.Aryldiazonium salts as dual precursors of aryl and arylhydrazine for the synthesis of polysubstituted pyrazoles.
In the aforementioned transformations,the radical-type interception can render aryldiazonium salts to act as dual synthons for the synthesis of important functionalized molecules.Moreover,the reactions involving ionic mechanism can be equally used to accomplish their versatility.In 2017,Tang and co-workers established an elegant copper(I)-promoted carboamination cascade reaction for the construction of 1,2,3-triazoles based on the employment of vinyl azides and aryldiazonium salts as the starting materials (Scheme 48) [82].Functionally diverse triaryl-substituted 1,2,3-triazoles could be gainedviaa difunctionalization ofβ-carbon of vinyl azides in satisfactory yields.It was identified that an addition of proper amount of 2,6-di-tert-butyl-4-methylphenol (BHT)could apparently increase the yield,implying that BHT might provide an appropriate acidic reaction system to ensure the occurrence of diazonium salts decomposition,meanwhile weakening the oxidation of Cu(I) by oxygen from air to some extent.The formation of aryl cation under the acidic conditions was assumed to be the first step.Then the aryl cation attacked the copper-chelated complex246resulting from vinyl azide,followed by the elimination of N2to generate an imine intermediate248.Alternatively,the 2H-azirine generated from vinyl azideviareleasing N2went through a ring-opening reaction with aryl cation to give the intermediate248.The tautomerization of248gave rise to the enamine248′,which underwent an addition with aryldiazonium cation to provide the complex249.The subsequent isomerization and reductive elimination enabled the formation of finalN2-substituted 1,2,3-triazole product244.

Scheme 48.Aryldiazonium salts as dual precursors of aryl and arylhydrazine for the synthesis of triaryl-substituted 1,2,3-triazoles.
7.Rongalite
Rongalite (sodium hydroxymethanesulfinate dihydrate),an adduct of formaldehyde and sulfur dioxide,is an abundant and cheap industrial product,which has been widely applied in the rubber,sugar,dye,and pharmaceutical industries [83].Owing to possessing both electrophilic and nucleophilic properties,rongalite is likely to react with amphiphilic reagents.To date,rongalite has been proven to be a practical and economical reagent for the preparation of sulfones under mild conditions,since it can serve well as the SO22-equivalent [84,85].Also,rongalite can be served as an ingenious C1 building blockviathe release of formaldehyde for the assembly of heterocycles (Scheme 49) [84].Moreover,rongalite can be used as a mediator to initiate radical reactions[86,87].Most recently,it has been proposed that rongalite can also act as a masked proton source [88].These rich reactivities render rongalite to become an emerging versatile reagent in synthetic chemistry.
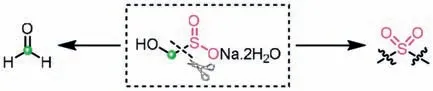
Scheme 49.Rongalite as the donors of SO22- and formaldehyde via the cleavage of C-S bond.
In this realm,Wu’s group made a conspicuous progress.In 2020,this group reported a novel and effective copper-catalyzed 1-thiaflavanone sulfones synthesis based on the utilization of rongalite as a safe and economic sulfone source (Scheme 50) [89].This reaction was performed in CH3CN with 1,10-phenanthroline as the ligand.Probably due to the poor solubility of rongalite in CH3CN,the addition of tetrabutyl ammonium salt was very imperative to increase the reaction yield.This sulfonylation showed a wide substrate scope,in which 2′-halogenchalcones bearing various alkyl substituents all smoothly proceeded,generating the corresponding products in generally good yields.The transformation was initiated by the coordination of 2′-halogenchalcone to copper catalyst to generate the intermediate253.Then a thia-Michael addition proceeded to give the intermediate254,which subsequently underwent the C-S bond coupling to provide the final product through the intermediate255with the release of formaldehyde.This protocol provided a simple and clean method for the synthesis of 1-thiaflavanone sulfones,avoiding the employment of toxic SO2gas as sulfone source.
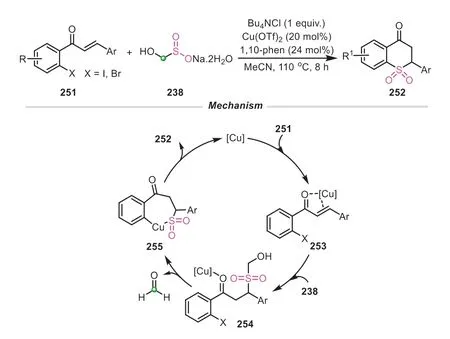
Scheme 50.Rongalite as SO22- donor for the synthesis of 1-thiaflavanone sulfones.
A similar strategy was also applied in the synthesis ofNaminosulfonamides by the same group (Scheme 51) [86].In the transformation,two molecules of aryldiazonium tetrafluoroborates were combined with rongalite,serving as aryl radical and amine source.Rongalite also played multiple roles,acting as the SO2surrogate,radical initiator and reduction reagent.The reaction involved a radical pathway.At first,rongalite decomposed into a HSO2-anion and formaldehyde.The generated HSO2-would assist one molecule of aryldiazonium cation to release nitrogen,generating aryl radical and sulfoxylate anion radical.The subsequent combination of the two radicals gave the crucial arylsulfinate257,which would attack another aryldiazonium salt,furnishing the sulfonyl diazo intermediate258.And the final reduction of258afforded the corresponding sulfonyl hydrazide256.Notably,the role of rongalite as the reductant was also further confirmed by the control experiments.
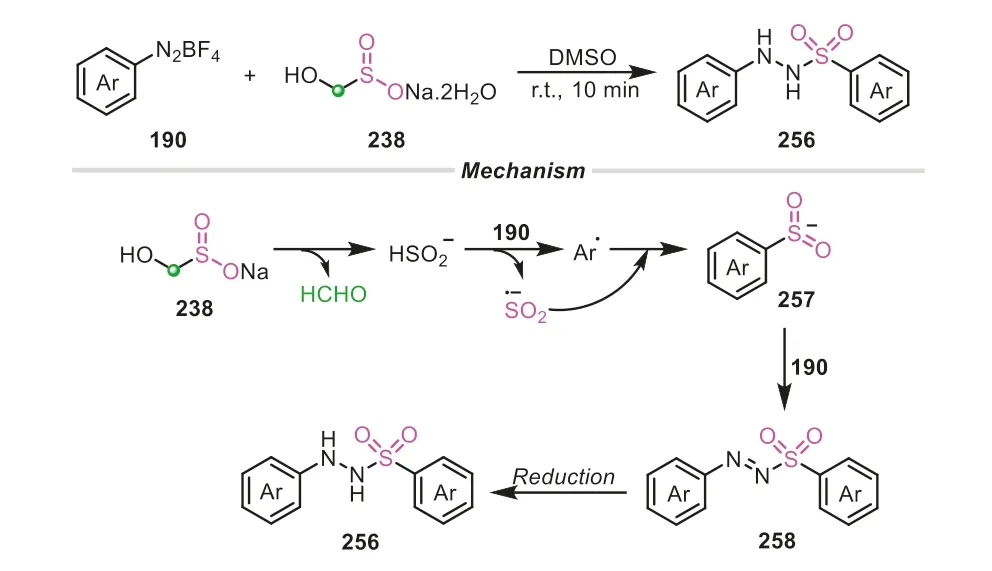
Scheme 51.Rongalite as SO22- donor for the synthesis of N-aminosulfonamides.
Rongalite is also used as the smart C1 subunit for the construction of valuable heterocyclic scaffolds.In 2016,this group firstly disclosed the utilization of rongalite as the formaldehyde source,which can be applied in the construction of furan ring (Scheme 52)[90].The reaction was performed in DMSO under the catalysis of I2/Cu(II),in which rongalite and aryl methyl ketones accomplished double C-S bond cleavages and triple C(sp3)-H functionalization reaction in one pot.The mechanistic study revealed that39reacted with molecular iodine to result inα-iodoacetophenone43,which was subsequently combined with dimethyl sulfide (DMS) to obtain the intermediate260.Meanwhile,formaldehydeinsituresulting from rongalite,was intercepted by260to deliver intermediate261in the presence of Cu(NO3)2·3H2O as Lewis acid.Then the combination of intermediate260and261proceeded to yield the intermediate263.

Scheme 52.Rongalite as the formaldehyde donor for the synthesis of furan derivatives.
Afterwards,263went through a consecutive isomerization/aromatization to afford the final 2,4,5-trisubstituted furan product.In the subsequent investigation,they further expanded the protocol for the preparation of C3-sulfenylated chromones(Scheme 53) [91].
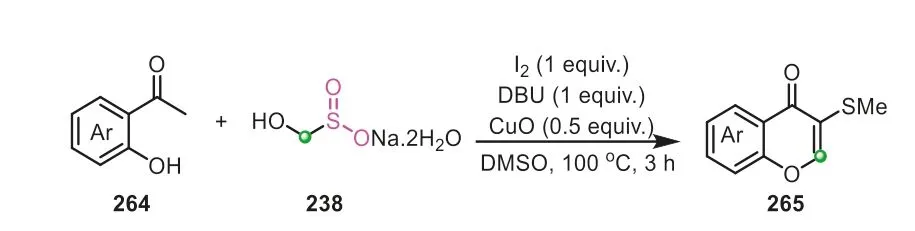
Scheme 53.Rongalite as formaldehyde donor for the synthesis of C3-sulfenylated chromones.
In 2022,Kokatlaetal.employed rongalite as a double C1 unit donor for the C(sp3)-H functionalization of 2-oxindoles(Scheme 54) [92].In addition,rongalite functioned as a hydridefree reducing agent in the transformation.Likewise,theinsitugenerated formaldehyde from rongalite underwent an Aldo condensation with indolin-2-one266to generate 3-methyleneindolin-2-one,which was followed by a reduction process with the help of rongalite,leading to 3-methylation of indolin-2-ones.The subsequent second aldol reaction occurred to further give 3-(hydroxymethyl)-3-methylindolin-2-one267.Notably,the reduction role of rongalite was also confirmed by the reaction of 3-methyleneindolin-2-one and rongalite under standard conditions.

Scheme 54.Rongalite as double C1 donors for the C-H functionalization of 2-oxindoles.
With the increasing exploration of rongalite as a highly reactive reagent,it has been emerging that employing rongalite as both C1 unit and sulfone equivalent concurrently converges into the final scaffolds in an atomically economical manner,albeit with a large challenge.In 2022,Wu and co-workers developed a concise and effective sulfonylmethylation strategy for sulfoxonium ylide (Scheme 55) [87].In contrast with traditional sulfonylmethylation methods by utilizing prefunctionalized sulfonylmethylating reagents,this protocol provided a novel separate-embedding sulfonylmethylation method by directly utilizing low toxic and readily available rongalite as versatile reagent under metal-free conditions,achieving this goal expediently and rapidly in one step.Mechanistic investigations revealed that the reaction involved a radical tandem process.A combination of sulfoxonium ylide and rongalite underwent a dehydration to generate the methylation species270,which was then reduced by rongalite or SO22-to afford theα,βunsaturated acetophenone271.At the same time,the interaction of rongalite and aryldiazonium salt provided the arylsulfinate257.With the aid of H+,the intermediate257further went through a thia-Michael addition with271to give the corresponding product269.
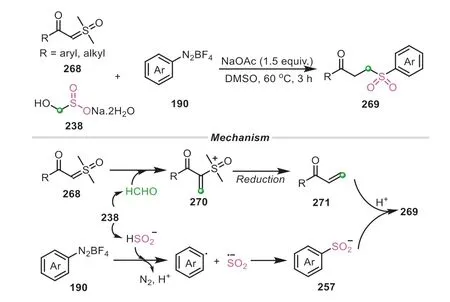
Scheme 55.Rongalite as C1 unit and sulfone source for the sulfonylmethylation.
In the same year,a new protocol using rongalite as triple C1 synthons and sulfone source to construct tetrahydro-2H-thiopyran 1,1-dioxide ring was established by the same group (Scheme 56)[93].Distinguished from the last strategy,two molecules of sulfoxonium ylides participated in the transformation,in which three C1 units were introduced into the six-membered ring framework.As shown in Scheme 56,α,β-unsaturated acetophenone was firstly generatedviathe condensation of sulfoxonium ylide with the formaldehyde from rongalite,followed by twice thia-Michael additions,giving the linear intermediate273.After that,an Aldo condensation on the intermediate273occurred to increase the carbon chain,which was subsequently accompanied by an intramolecular Michael addition to deliver the final six-membered heterocyclic product272.The proposed mechanism implied that two different carbon-increasing models were demonstrated in the transformation,in which rongalite firstly functionalized as a tethered C-S synthon,and was subsequently performed as a simple methylene to be incorporated into the products.
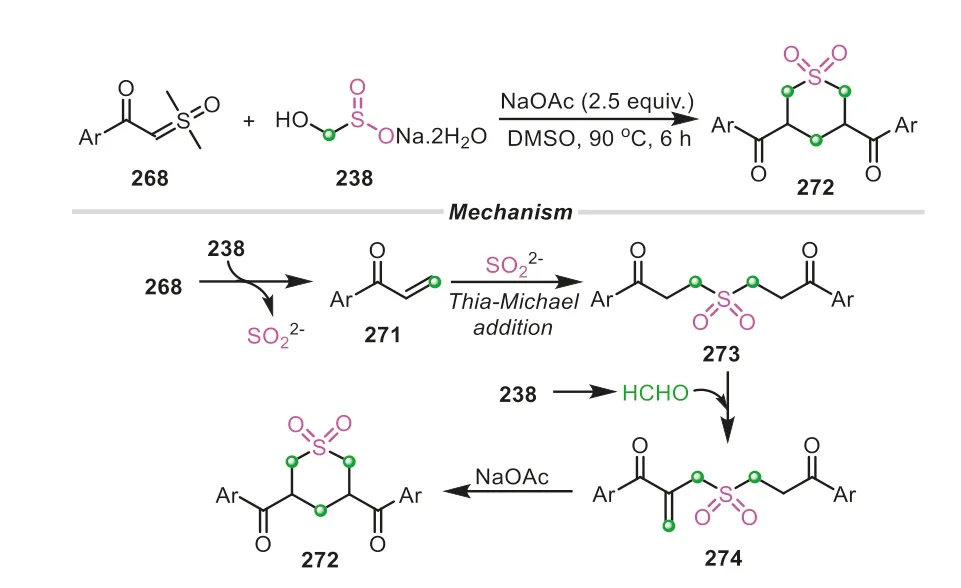
Scheme 56.Rongalite as triple C1 donors and sulfone source for the construction of tetrahydro-2H-thiopyran 1,1-dioxide ring.
8.Halodifluoromethyl compounds
Halodifluoromethyl compounds,especially for halodifluoroacetates (halo=Br,Cl or I),are an important class of fluorinecontaining reagents,which have been extensively investigated as(gem)-difluoroalkylative reagents [94,95] and difluorocarbene synthons [96-98],viaC-X bond or (and) C-R bond cleavage controlled by different reaction conditions (Scheme 57).It must be mentioned that difluorocarbene is a singlet state one having three sp2hybrid orbitals and an emptyp-orbital,rendering it intrinsically electrophilic,which makes it different from other carbene species,even other dihalocarbenes [99].Intriguingly,it has been common that halodifluoroacetates undergo a quadruple cleavage to result in a novel C1 source.Besides,they can also provide the fluorine source directly.Of note,these diverse reactivities can be also performed in the same reaction simultaneously.Thus these functions have gradually enabled halodifluoroacetates to be a new class of multifunctionalized reagents in the field of organic synthesis.

Scheme 57.Halodifluoromethyl compounds as versatile synthons via multiple bond scissions.
In the utilization of halodifluoromethyl compounds,Song’s group has made extensive investigations and achieved remarkable progress.In 2016,Song and co-workers reported a costeffective copper-catalyzed hydrofluoroacetylation reaction of arylacetylenes from cheap and readily available bromo-substituted difluoroacetates (Scheme 58) [100].This reaction showed a broad substrate scope and high stereoselectivity,providing diverse (E)-fluoroacetylated alkenes with good to excellent yields.Remarkably,alkynyl carboxylic acids were also proven to be the suitable substrates for this hydrofluoroacetylation,even revealing better stereoselectivities compared to the corresponding alkynes.The mechanism investigation suggested that the reaction passed through a radical pathway,in which (phenylethynyl)copper might be a crucial intermediate.In addition,B2pin2also played a vital role,maybe serving as the reductant in the reaction.
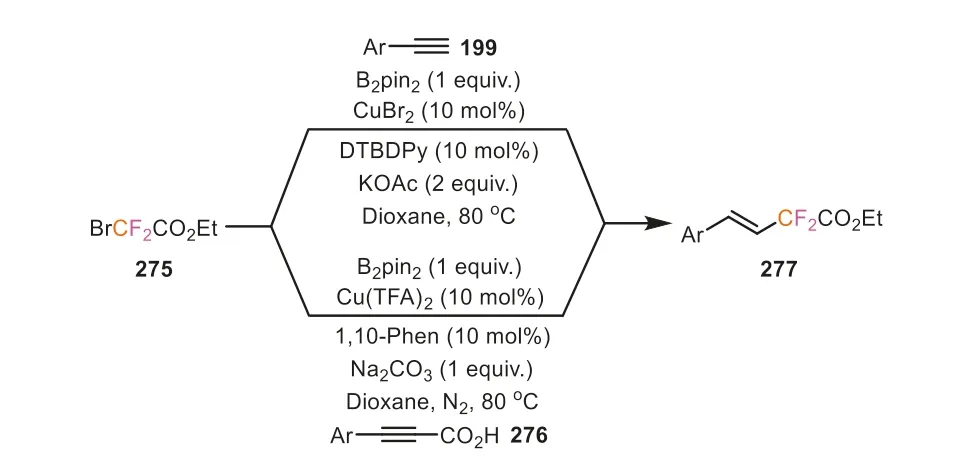
Scheme 58.Ethyl bromodifluoroacetate as difluoroalkylating reagent for the hydrofluoroacetylation of arylacetylenes and alkynyl carboxylic acids.
In the next year,this group also employed the Cu(II)/B2pin2-catalytic system for the difluoroacetylation of aniline,preparing an important class of 3,3-difluoro-2-oxindole derivativesviaa successive C-H activation/intramolecular amidation process (Scheme 59)[101].The reaction showed good regioselectivity,whether primary,secondary or tertiary anilines regiospecifically provided theorthodifluoroacetylated products.And the first two cases could further undergo intramolecular amidation,generating 3,3-difluoro-2-oxindole productsviaa one-pot strategy.Most importantly,this strategy can be applied as a key step for the preparation of an important agent,EP3receptor antagonist,obviously shortening the steps of traditional approach.The above two transformations elegantly demonstrated the use of BrCF2COOEt as radical precursors through the cleavage of single C-Br bond.

Scheme 59.Ethyl bromodifluoroacetate as difluoroacetylation reagent for the synthesis of 3,3-difluoro-2-oxindoles.
In their subsequent investigations,they also found that ethyl bromodifluoroacetate could undergo an intriguing quadruple cleavage,including one C-Br bond,two C-F bonds and one C-COOEt bond concurrently (Scheme 60) [102],leading to the formation of a novel C1 source,which could be captured by primary amines to form isocyanideinsitu,followed by a cross-coupling with 1,3-dicarbonyl compounds to accessβ-aminoenones.It is worth mentioning that a wide range of primary amines,such as aromatic amines,heteroaromatic amines,and aliphatic amines,were all compatible with this transformation,delivering the correspondingβ-aminoenone products in good to excellent yields.Most remarkably,tert-butyl amine and adamantan-1-amine that failed to deliver the targetβ-aminoenone products starting with the corresponding isocyanides as materials in the previous report,were also proven to be feasible for this protocol [103].The reaction mechanism was proposed in Scheme 60.Theinsitugenerated difluorocarbene from BrCF2CO2Et was attacked by primary amine to result in a labile281,which was subsequently converted into isocyanide in the presence of base.The following reaction of isocyanide and 1,3-dicarbonyl source delivered theβ-aminoenone product.
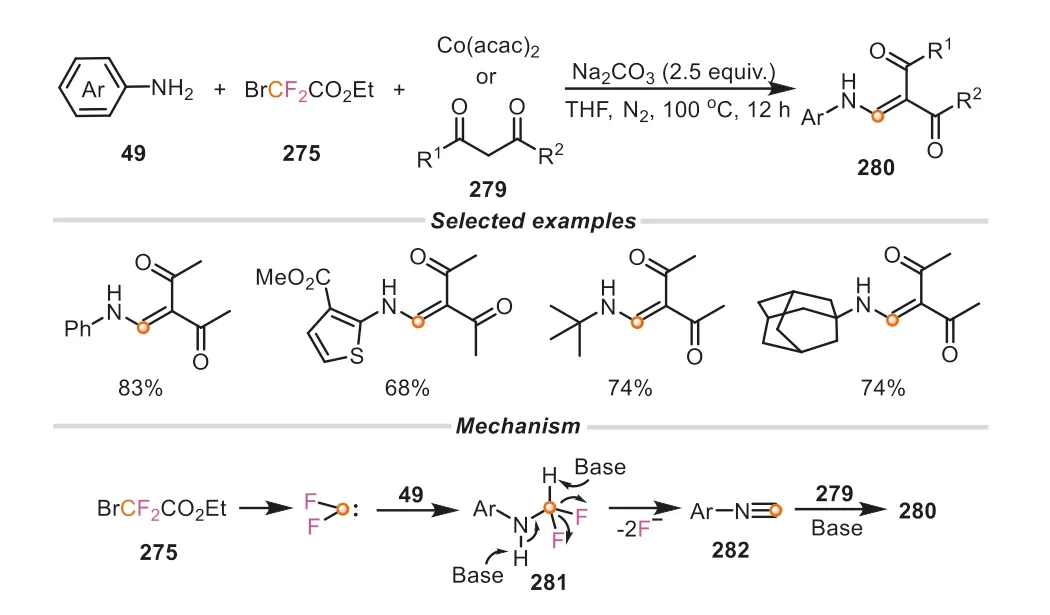
Scheme 60.Ethyl bromodifluoroacetate as C1 source for the synthesis of βaminoenones.
In the reaction between primary amines with BrCF2CO2Et,the formation of isocyanide is a key processviatwice C-F bond cleavage.Intriguingly,it was founded that the employment of secondary amines can lead to the assembly of various formamides,which was also disclosed by the same group (Scheme 61) [104].This reaction result was mainly explained that the second C-F bond cleavage was prevented,instead,other processes,such as hydrolysis or SNAr substitution process might happen.The formation of formamides was proposed as follows: the difluorocarbene species was generated under basic conditions,which was attacked by a secondary amine to afford the intermediate285.Subsequently,285underwent a self-attack by lone-pair electrons at nitrogen atom and produced an unstable intermediate286viaa defluorinative pathway.With the assistance of base,286was rapidly transformed into287or288by SNAr substitution (path a) or nucleophilic addition (path b),ultimately providing the formamide product.In this transformation,BrCF2COOEt served as a formylation reagent.In contrast with the last example,it can be easily concluded that substrate types perform a huge effect on reaction pathways.
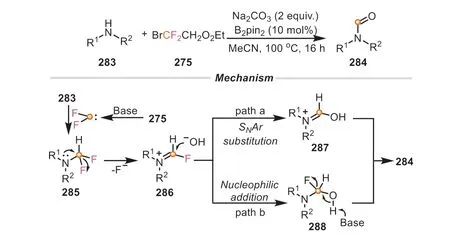
Scheme 61.Ethyl bromodifluoroacetate as formylation reagent for the synthesis of formamides.
1,3,5-Triazines belong to an advantageous and important class of structural motifs,found in numerous natural products and pharmaceutical molecules [105].Moreover,1,3,5-triazines can be regarded as promising ligands in organic synthesis [106].In light of their indisputable importance and widespread applications,Songetal.established a simple and fascinating cyclization reaction of amidines with ethyl bromodifluoroacetate for the preparation of symmetric and unsymmetric 2,4-disubstituted-1,3,5-triazines in the presence of baseviamultiple C-N bonds formation,in which ethyl bromodifluoroacetate acted as a unique C1 source(Scheme 62) [107].Theinsituformed difluorocarbene with the assistance of Na2CO3reacted with amidine to afford the intermediate291,which was further converted to imine intermediate292by the C-F bond scission.Subsequently,the reactive292was captured by another molecule of amidine to produce the intermediate293,followed by an intramolecular nucleophilic addition to deliver the cyclized product 1,3,5-triazine.These examples fully demonstrated the versatility of halodifluoromethyl compounds as C1 source in constructing structurally diverse organic molecules.
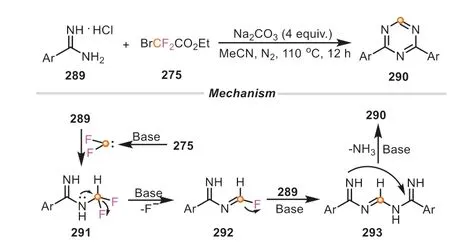
Scheme 62.Ethyl bromodifluoroacetate as C1 source for the synthesis of 1,3,5-triazines.
In some cases,ethyl bromodifluoroacetate can perform dual functions,such as acting as C1 source and difluoroalkylation reagent synchronously.In 2018,Song and co-workers for the first time disclosed the dual roles of ethyl bromodifluoroacetate by employingortho-substituted arylamines as the coupling partners(Scheme 63) [108].The reaction of 2-arylaniline with BrCF2COOEt yielded 6-difluoroacetate phenanthridines in good yields,while the combination of BrCF2COOEt and 2-amino thioanisoles led to the formation of various 2-difluoroacetate benzothiazoles with good functional group tolerance.It should be pointed out that 30 mol%of B2pin2was essential for promoting the latter one.In these transformations,the use of these primary anilines all afforded the corresponding isocyanide intermediates in the presence of base,which subsequently went through a cascade annulation and difluoroalkylation,eventually furnishing the final product.
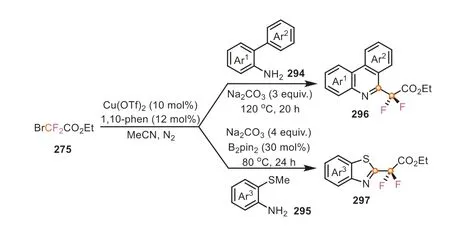
Scheme 63.Ethyl bromodifluoroacetate as C1 and difluoroalkylating reagents for the synthesis of fluorine-containing heterocycles.
In 2022,the same group also successfully unveiled a novel difunctionalized roles of BrCF2COOEt as C1 and F1 reagent for the synthesis of 3-fluorinated oxindoles (Scheme 64) [109].This transformation featured an unusual reaction pathway,in which thein situformed active species difluorocarbene initially interacted with the amino fragment of 2-aminoarylketone,leading to the ammonium salt300.Next,theα-C of300was attacked by the bromine anion to afford indoline intermediate301.The subsequent scission of one C-F bond rendered the formation of epoxide intermediate302.The final 1,2-fluorine migration occurred to yield 3-fluorinated oxindole product.The reaction was characterized by several other advantages,such as simple operation,broad substrate scope,as well as high efficiency.
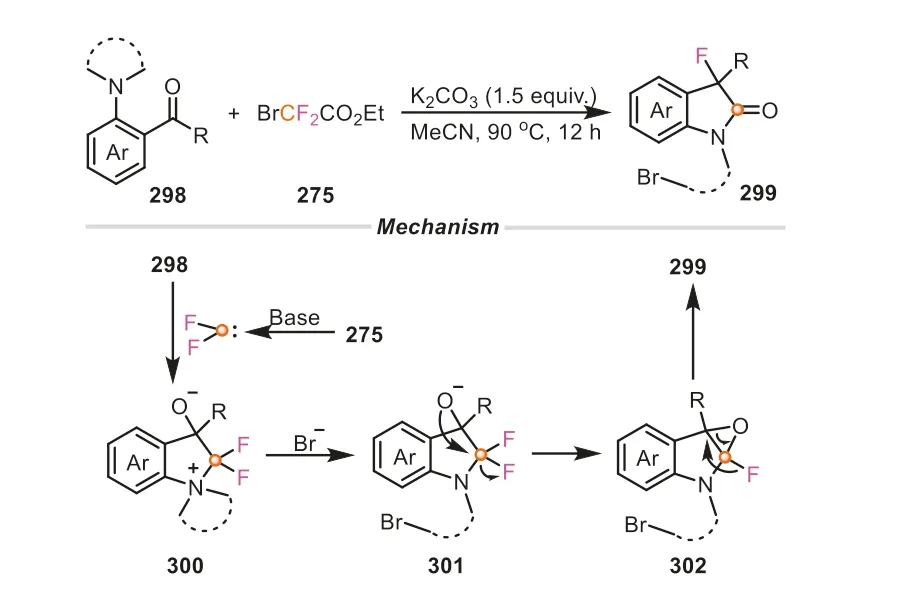
Scheme 64.Ethyl bromodifluoroacetate as C1 and F1 reagents for the synthesis of 3-fluorinated oxindoles.
Apart from these,halodifluoromethyl compounds can be engaged as the dual fluorine sources to be introduced into the final target products.Using BrCF2COOEt as the different fluorines,they provided a concise and elegant strategy to achieve 3-(2,2-difluoroethyl)-2-fluoroindoles fromo-aminostyrenes (Scheme 65)[110].Throughout the whole cascade reaction process,two molecules of difluorocarbene species were captured successively byo-aminostyrenes,enabling that two different fluorine motifs could be incorporated into the productsviaforging one C-N and two C-C bonds concurrently.This reaction was performed in CH3CN under mild conditions,without any transition metal catalyst and additive.Most remarkably,the strategy also showed great potential in the late-stage modifications of complex natural products,which rendered the methodology more attractive and practical.Overall,these aforementioned results clearly revealed the superiorities of BrCF2COOEt as versatile synthons in constructing prevalentN-heterocycles.
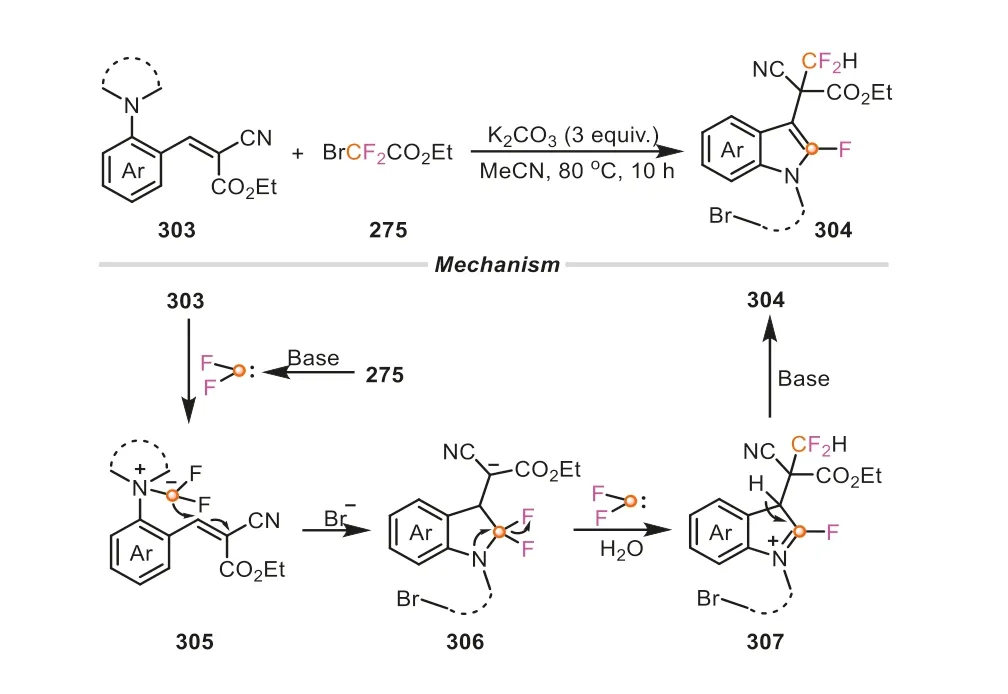
Scheme 65.Ethyl bromodifluoroacetate as C1 and dual fluorine sources for the synthesis of 3-(2,2-difluoroethyl)-2-fluoroindoles.
9.Conclusion and outlook
The rapid assembly of structurally complex and diverse molecules from simple starting materials is one of the major goals in modern organic synthesis.In this regard,the employment of versatile reagents is an elegant and efficient strategy to accomplish this target.By choosing suitable counterparts and adjusting reaction conditions,versatile reagents can be used as controllable and flexible building blocks for the divergent synthesis of important organic molecules.Over the past decade,a variety of multifunctional reagents have been developed.However,there are large limitations in terms of the practicalities and universalities.In this review,we prefer to focus on the use of some simple and readily available versatile reagents,mainly including atropaldehyde acetal,acetophenone,vinylene carbonate,vinyl azide,aryldiazonium salts,rongalite,halodifluoromethyl compounds.Based on the rational design of reaction pathways,these energetic reagents have been successfully applied to construct valuable skeletons by specific bond-cleaving and bond-forming modes.Moreover,these versatile reagents can also play dual roles simultaneously in the same reaction,in which their different reactivities are converged into the final target products.Such strategies can not only offer more possibilities for the synthesis of several useful products,but also minimize the occurrence of some side reactions by lessening the varieties of materials.
Although great progress has been achieved in the development and utilization of versatile reagents,some challenges are still encountered.Firstly,the variety of common and practical versatile reagents is far from satisfactory,thus continuous efforts should be devoted to developing novel renewable multifunctional reagents.In addition,the reactivities of partial versatile reagents are not explored completely,and the random combinations of their diverse reactivities are still not fully achieved.In some cases,the reaction conditions are harsh,such as performing at extreme temperatures or using large excess toxic solvents and expensive additives,which are inconsistent with the pursuit of green and sustainable chemistry.The above-mentioned problems are remaining to be solved by organic chemists.We anticipate that this overview can encourage and stimulate chemists to design more interesting and useful reactions,thus enriching the reactivities of versatile reagents.Meanwhile,we also hope that these reagents serve as flexible and controllable building blocks to deliver more valuable molecules in the future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No.22072049),National Key Research and Development Project (No.2022YFE0124100),and Major Special Projects of Science and Technology of Ordos (No.2022EEDSKJZDZX003).Program for HUST Academic Frontier Youth Team (No.2019QYTD06) is also acknowledged.Chen is also grateful for the financial support from China Scholarship Council (CSC).
 Chinese Chemical Letters2024年2期
Chinese Chemical Letters2024年2期
- Chinese Chemical Letters的其它文章
- The 3rd Xihua Chemistry and Biomedicine Forum
- Professor Hualiang Jiang: A tribute to an esteemed visionary chemist and pharmacist
- Recent advances in visible light-mediated chemical transformations of enaminones
- Development of porphyrin-based fluorescent sensors and sensor arrays for saccharide recognition
- Synthetic host-guest pairs as novel bioorthogonal tools for pre-targeting☆
- Functional nanoemulsions: Controllable low-energy nanoemulsification and advanced biomedical application