
Photo-induced 1,2-aryl migration of 2-chloro-1-arylpropanone under batch and flow conditions: Rapid,scalable and sustainable approach to 2-arylpropionic acids
2024-04-05 02:28QiaoLiMinjieLiuMeifenJiangLiWanYingtangNingFenErChena
Chinese Chemical Letters 2024年2期
Qiao Li ,Minjie Liu ,Meifen Jiang ,Li Wan ,Yingtang Ning,* ,Fen-Er Chena,,d,*
a College of Chemical Engineering,Zhejiang University of Technology,Hangzhou 310014,China
b Engineering Center of Catalysis and Synthesis for Chiral Molecules,Fudan University,Shanghai 200433,China
c Shanghai Engineering Center of Industrial Catalysis for Chiral Drugs,Shanghai 200433,China
d Institute of Pharmaceutical Science and Technology,Zhejiang University of Technology,Hangzhou 310014,China
Keywords: Green chemistry Flow chemistry 2-Arylpropionic acids Photo-Favorskii reaction
ABSTRACT We report the photo-mediated 1,2-aryl migration of 2-chloro-1-arylpropanones to 2-arylpropionic acids using HCOONa as an acid scavenger.This pragmatically focused study obviates the multiple-step sequence in the industrially employed,ZnO-promoted rearrangement strategy,and offers rapid access to various 2-arylpropionic acids under environmentally friendly conditions.Furthermore,the successful transfer of this batch photochemistry to a continuous flow platform led to improved scalability and enabled the gramscale synthesis of loxoprofen.
2-Arylpropionic acids are prevalent in bioactive molecules and marketed pharmaceuticals [1].For example,they represent an important class of non-steroidal anti-inflammatory drugs (NSAIDs),including ibuprofen [2] and loxoprofen [3].Hence,considerable effort has been dedicated to developing methodologies that enable efficient and sustainable synthesis of such structures [4].Although several approaches are available,such as methylation of aryl acetic acid derivatives [5],hydrolysis of corresponding nitriles [6],and transition metal-catalyzed carbonylation reactions [7],they are often plagued with the low availability of substrates,the handling of toxic reagents,and the harsh reaction conditions.
The 1,2-aryl migration ofα-substituted alkyl aryl ketones is a practical strategy for preparing 2-arylpropionic acids [8,9].The ZnO-promoted migration ofα-chloro ketals is adapted as a key step for the manufacture of ibuprofen in China at>8000 tons of annual production capacity (Scheme 1B,route 1) [10,11].On the other hand,the need to acetalize the carbonyl group before the migration and the subsequent hydrolysis procedures has stimulated further advances in rearrangement protocols that can bypass this tedious sequence.Representative examples include the oxidative migration of propiophenone derivatives (route 2)[12-15] and the photo-initiated Favorskii rearrangement ofαchloroketones (route 3).In particular,the photorearrangement process has attracted continuous attention for its cost efficiency and adhere to the principles of green chemistry [16,17].To this end,Sonawane and others made significant contributions to the photochemical rearrangement of 2-chloro-1-arylpropanones [18-21].Baxendale recently applied this chemistry to the flow synthesis of ibuprofen using a continuous photochemistry platform for enhanced photon delivery [22].However,all the existing reports employed (over)stoichiometric propylene oxide as an acid scavenger,which is acutely toxic and carcinogenic with high vapor pressure(vp=445 mmHg at 20°C).
To design a more efficient photo-rearrangement protocol,we pursued a non-hazardous alternative to propylene oxide.Discussions based on the reaction mechanism suggest that an ideal acid scavenger should be a mild base and poor electron donor(Scheme 2) [23-25].Mechanistic investigations of photo-Favorskii rearrangements of the closely related,p-hydroxyphenacyl derived3suggested the generation of triplet33*under light irradiation[26,27].Adiabatic formation of the triplet biradical34followed by3I →1I ISC furnished the Dewar zwitterion5[28,29],where the deprotonation of33*is likely assisted by general acid and general base catalysis of water [30,31].For1awithout ap-OH group,the absence of a phenolic proton should shut down such a reaction channel.Instead,deprotonation could occur with strong bases at other acidic sites,such as theα-position of the carbonyl group substituted with a Cl atom (Scheme 2B and Scheme S1 in Supporting information) and the acidic proton in product2a[32-34].Moreover,an additive could act as a hydrogen atom donor or an electron donor,facilitating the photoreduction pathways to generate8a[35-39].Bearing these considerations,we report the photo-Favorskii rearrangement of1using HCOONa as an acid scavenger.The current protocol was further transferred to a continuous-flow platform,leading to improved photon delivery,reproducibility,and scalability [40-46].
We embarked on initial optimization efforts using the photoinduced rearrangement of 2-chloro-1-(p-tolyl)propan-1-one (1a) as a model reaction (Table 1).The desired acid2awas obtained in 50% yield at 55°C under irradiation of 365 nm in a mixed solution of acetone-water (w/w=9:1) (entry 1),and the yield of2aincreased in the presence of propylene oxide (entry 2).The addition of mild bases led to decreased yields of2a(Table S1 in Supporting information).For example,when triethylamine was used,the desired2awas obtained in a marginal 13% yield (Table 1,entry 3),with the dehalogenated7adetected as the major byproduct (62%yield).The reaction employing pyridine delivered2ain a 79% yield(entry 4),which is comparable to the conditions with propylene oxide (entry 2).These results suggested that the reaction yields were inversely related to the basicity and reduction ability of additives,which encouraged us to examine the effect of carboxylate salts.While sodium acetate was less satisfying (entry 5),sodium formate [47] proved to be an effective replacement for the hazardous propylene oxide (entry 6).
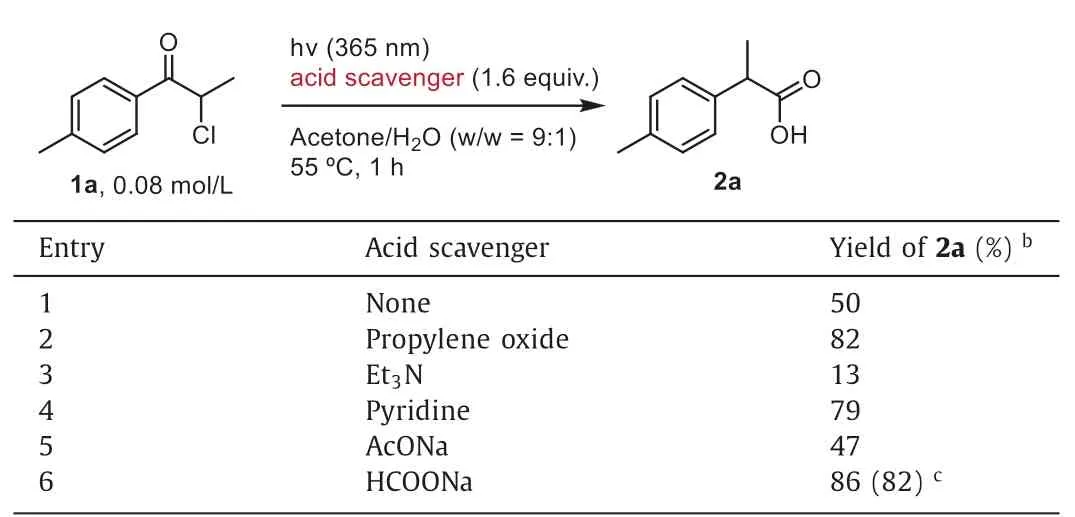
Table 1Screen of acid scavengers.a
Further evaluations of other reaction conditions were performed with HCOONa (Table 2 and Table S1).The reaction operated under the wavelength range of 335-385 nm (entries 2 and 3,see Figs.S1 and S2 in Supporting information for details of the UV-vis absorbance spectra).Increasing the equivalence of HCOONa showed little improvement in the reaction yield in batch (entry 4).Acetonitrile (ε=37.5) can also be used as a solvent (entry 5),but other solvents,such as THF (ε=7.6) and DMF (ε=2.3),gave a mixture of2aand7a(entry 6) [48].Lowering the reaction temperature to 45 °C led to a slightly compromised yield (80%,entry 7).The rapid kinetics of the photo-induced migration is well illustrated in entry 8.Irradiation of1afor 5 min yielded2a,and a trace amount of1awas recovered (89% conversion).

Table 2Optimization of reaction conditions in batch.a
Next,we sought to address the scalability issue inherent to photochemical transformations under batch conditions.Transfer-ring a photochemical reaction to a continuous-flow platform offers many advantages,such as ease of scaling up,enhanced mass transfer,and more homogeneous irradiation.We began our investigations of the continuous-flow reaction using a homemade photoreactor with a reaction unit featuring coiled FEP tubing (0.8 mm ID,4.0 mL).The coil was placed in a water bath to control the reaction temperature (Fig.S2).The limited solubility of HCOONa and NaCl (as a byproduct) resulted in solid formation upon extended irradiation in batch reactions.In flow,mixing a solution of1ain neat acetone (0.086 mol/L) and HCOONa in water (1.5 mol/L) prevented precipitation (Table 3).A quick screen of flow rates (entries 1-4) identified the optimal conditions (entry 3),and2awas successfully obtained in 85%-87% yields within 4 min using the flow protocol (entry 3).Further reducing the residence time led to decreased yield (entry 5).Similar to batch reactions,acetonitrile could serve as an optional solvent (entry 6).We also examined the effect of pyridine as a basic additive in flow,and2awas obtained in 80% yield (entry 7).
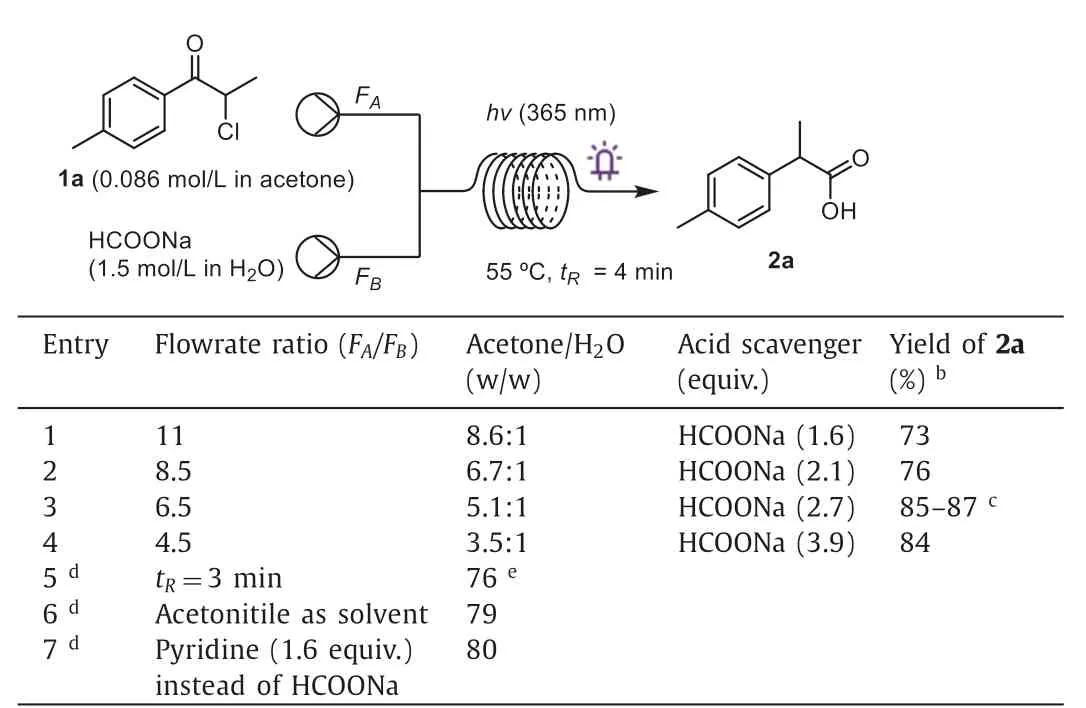
Table 3Optimization of reaction conditions in flow.a
With optimized conditions in hand,we explored the generality of the photochemical aryl migration module in batch and flow(Scheme 3A).A collection ofpara-alkyl substituted derivatives underwent the rearrangement with good yields of the desired products (2b-2f).Notably,both the batch and flow reactions of1dproceeded smoothly to yield ibuprofen2dwithout acetalizationhydrolysis steps.To our delight,the reaction tolerated the ketone functional group at a remote position (2eand2f),and loxoprofen2fwas isolated in 69% and 79% yields in batch and flow under the established conditions.Also,m-Me substituted1gwas an effective substrate (2g,81% yield in flow),buto-Me substitution led to undesired Norrish type II reactions (videinfra).
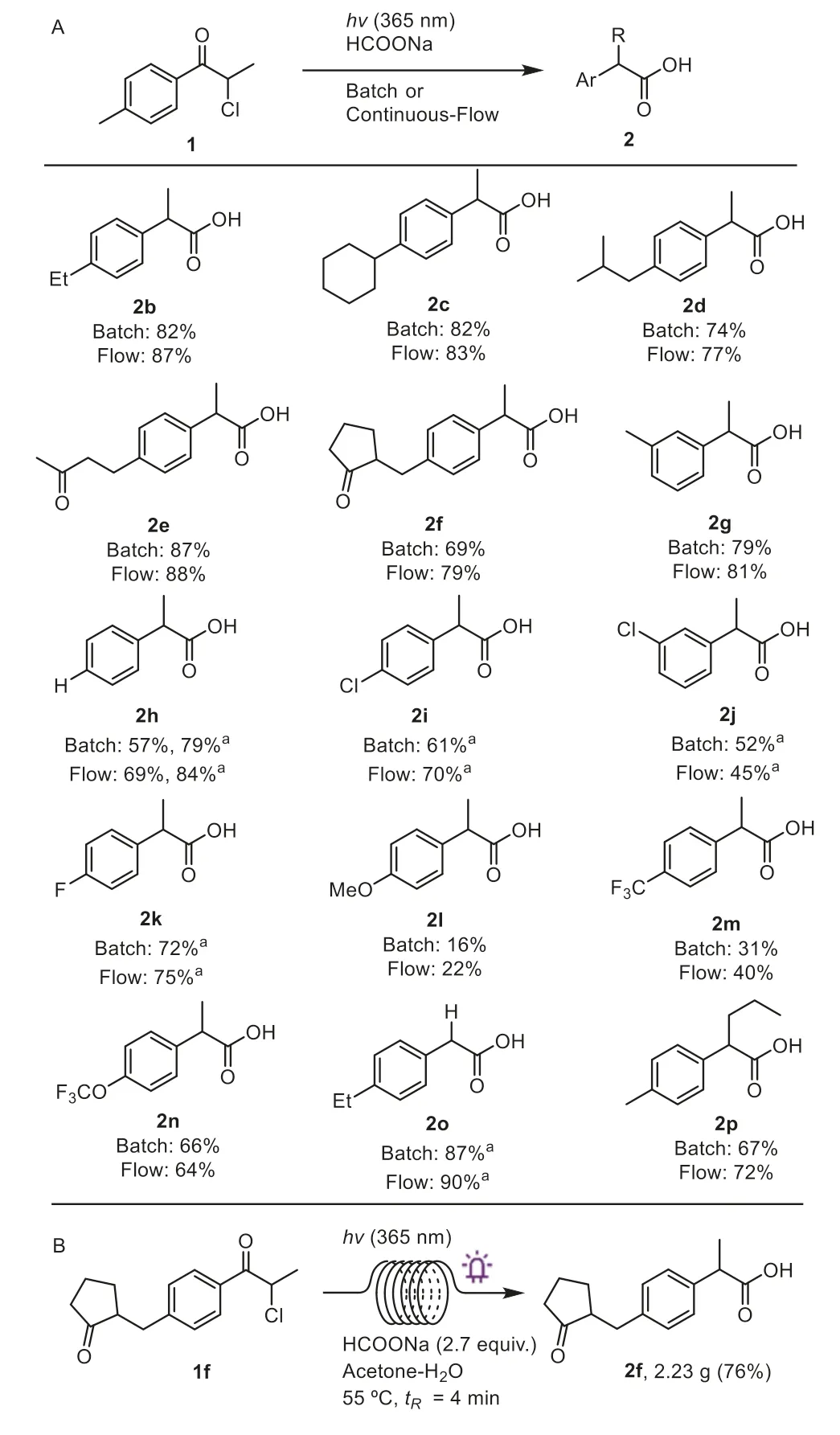
Scheme 3.Substrate scope (A) and scale-up synthesis of loxoprofen (B).Batch reactions were performed with 0.4 mmol 1 at 55°C for 1 h.Flow reactions were performed with 2.0 mmol of 1 at 55°C with tR=4 min.All reported yields are based on isolated 2.a MeCN was used as solvent.
In general,substrates without electron-donating alkyl substituents reacted with better yields in polar MeCN (ε=37.5) than in acetone (ε=20.5).As such,2-phenylpropanoic acid (2h) was obtained in satisfying yields in MeCN (84% in flow).Similarly,halogen substitution was accommodated to provide2i-2kwith additional functional handles.In addition,products withp-OMe (2l)andp-CF3(2m) were obtained in poor yields along with photolysis products.Trifluoromethoxy substituted2nwas obtained in a moderate 66% yield,indicating the window for the electronic properties of substituents.Structural variations of the substrates delivered2oand2pwith differentα-alkyl substituents.
The reaction setup is straightforward and requires no precautions for dry solvents and air.These features,coupled with the environmental benefits of photochemical transformations,demonstrate the suitability of this method for the decagram synthesis of NSAIDs.Specifically,multigram quantities of1freacted well under standard conditions,and loxoprofen (2f,76%) was isolated by recrystallization (Scheme 3B).
Additional observations concerning the detailed reaction pathway under the current conditions are shown in Scheme 4.We performed the reactions with triethylamine and sodium formate in D2O and observed no evidence of deuterium incorporation in2a.On the other hand,deuterium exchange was feasible for1ain the dark (Scheme S1).Accordingly,deprotonation of1aand related intermediates is not involved during the desired photochemical pathway.The attenuated yields of2land2mwere attributed to the presence ofα-cleavage pathways [49],where photolysis of2land2mcompeted with the desired 1,2-aryl migration to yield the Norrish Type I products12land12m.Similarly,a hydrogen atom abstraction-cyclization mechanism was operative foro-Me substituted2q[50,51],as evidenced by the formation of13q.Overall,these results are consistent with the reaction manifold [20] depicted in Scheme 2C.
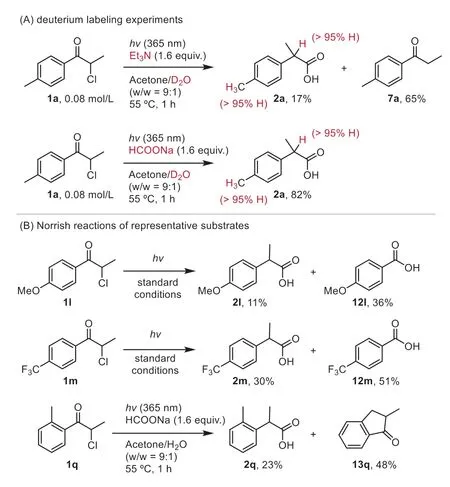
Scheme 4.Experimental results of detailed reaction pathways.
In summary,the photo-Favorskii rearrangement ofαchloroketones to produce 2-arylpropionic acids was achieved by using HCOONa as an acid scavenger.Incorporating the continuousflow setup avoided the issues associated with batch-mode photochemistry,showing more reproducible and scalable results.This method was compatible with the multigram synthesis of NSAIDs,and we expect that the scalability and the experimental ease of this photo-induced transformation will lead to its extensive use.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
Financial support from the National Natural Science Foundation of China (No.22077018) is gratefully acknowledged.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108576.
 Chinese Chemical Letters2024年2期
Chinese Chemical Letters2024年2期
- Chinese Chemical Letters的其它文章
- The 3rd Xihua Chemistry and Biomedicine Forum
- Professor Hualiang Jiang: A tribute to an esteemed visionary chemist and pharmacist
- Recent advances in visible light-mediated chemical transformations of enaminones
- Development of porphyrin-based fluorescent sensors and sensor arrays for saccharide recognition
- Recent advances of versatile reagents as controllable building blocks in organic synthesis
- Synthetic host-guest pairs as novel bioorthogonal tools for pre-targeting☆