
A proteomic landscape of pharmacologic perturbations for functional relevance
2024-03-21 05:51ZhiweiLiuShngwenJingBingingHoShuyuXieYingluoLiuYuqiHungHengXuChengLuoMinHungMinjiTnJunYuXu
Zhiwei Liu , Shngwen Jing , Binging Ho , Shuyu Xie , Yingluo Liu , Yuqi Hung ,Heng Xu , Cheng Luo ,, Min Hung , Minji Tn ,,c,d, Jun-Yu Xu ,,c,*
a State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China
b School of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing, 210023, China
c Zhongshan Institute for Drug Discovery, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zhongshan, Guangdong, 528400, China
d State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, 210023, China
Keywords:Proteomics Drug Perturbation Drug target Drug combination
A B S T R A C T Pharmacological perturbation studies based on protein-level signatures are fundamental for drug discovery.In the present study,we used a mass spectrometry(MS)-based proteomic platform to profile the whole proteome of the breast cancer MCF7 cell line under stress induced by 78 bioactive compounds.The integrated analysis of perturbed signal abundance revealed the connectivity between phenotypic behaviors and molecular features in cancer cells.Our data showed functional relevance in exploring the novel pharmacological activity of phenolic xanthohumol,as well as the noncanonical targets of clinically approved tamoxifen, lovastatin, and their derivatives.Furthermore, the rational design of synergistic inhibition using a combination of histone methyltransferase and topoisomerase was identified based on their complementary drug fingerprints.This study provides rich resources for the proteomic landscape of drug responses for precision therapeutic medicine.
1.Introduction
Molecular profiling of drug-response phenotypes expands our knowledge of cancer pharmacology and provides guidance for precision medicine [1].Systematic molecular profiling is a useful approach to understand the mechanisms of action(MOA)of drugs in cancer cell lines[2].Currently,different layers of omics data are considered to reflect cell-line perturbation after compound treatment.These layers include genomic data at the DNA or messenger RNA (mRNA) level, as well as metabolomics and proteomics data.As end products encoded by genes,targets of small molecules,and direct executors of all biological functions,proteins are superior in acting as functional and proximal readouts that directly reveal drug actions [3].Genomic sequences are not disturbed during shortterm drug stimulation, which restricts their application in deciphering molecular mechanisms.Metabolites and transcripts do not provide a direct readout of the perturbagen response,limiting their ability to capture slight signal changes.In recent years,breakthroughs in mass spectrometry (MS)-based proteomic technology have promoted the technical feasibility of exploring unbiased proteome perturbations in drug responses.To date,large-scale proteomic drug responses as signature profiles and the integration of drug sensitivity in perturbed cancer cell lines have been reported[4-7].These preliminary studies explored the current understanding of potential biomarker and drug target discovery.However, some important drug types such as the immunomodulatory drugs (IMiDs) were not included in these studies.Recently, a proteome-wide atlas of drug MOA in the colon cancer cell line HCT116 has been reported[8];however,the cancer type of the cell lines dominates the molecular classification[9].Additionally,these databases have not been exhaustively utilized for their potential applications in pharmacological exploration, particularly for exploring novel drug MOA and predicting drug combinations.
To fill this gap, we used a Tandem Mass Tag (TMT)-based proteomic strategy to establish proteomic signature data for 78 distinct bioactive small molecules in the breast cancer epithelial MCF7 cell line.We then compared and analyzed the signature of the published proteomic data underlying compound treatment with our dataset.Based on the similarity of protein expression profiles, we uncovered novel mechanism or “off-target” effects of bioactive compounds.Furthermore, we revealed and validated a rational design for a drug combination strategy.Our results provide a resource for the systematic characterization of the molecular responses to drug treatments.These datasets can serve as functional proteomic platforms to facilitate drug discovery and inform new therapeutic approaches.
2.Experimental
2.1.Compounds
The compounds were obtained commercially or from the compound library of the Institute of Medicine, Chinese Academy of Sciences.The details of these compounds are listed in Table S1.
2.2.Cell lines
MCF7 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco, Waltham, MA, USA) with 2 mM L-glutamine(Amresco,Solon,OH,USA),10%fetal bovine serum(FBS;Gibco),and 100 units/mL of penicillin/streptomycin (Meilunbio,Dalian,China)and incubated at 37°C in 5% CO2.Cells were collected after drug treatment for 12 h.Three experimental periods were conducted,the details of which are listed in Table S1.
2.3.Quantitative proteomics
The cells were cultured in a 15-cm dish at approximately 90%confluency, washed with 1× phosphate-buffered saline (PBS,Meilunbio)and lysed using freshly prepared lysis buffer(8 M urea,100 mM NH4HCO3,and 2×protease inhibitor cocktail(Roche,Basel,Switzerland); pH 8.0).The cell lysate was incubated at 4°C for 30 min and then sonicated for 2 min with 30% energy (ultrasonic cell disruptor JY92-II, Ningbo Scientz Biotechnology Co., Ltd.,Ningbo, China).
For each sample,50 μg of proteins(determined by bicinchoninic acid assay) was reduced with 5 mM dithiothreitol (Sigma-Aldrich,St.Louis, MO, USA) for 30 min at 56°C.Cysteine was alkylated by incubating with 15 mM iodoacetamide (Sigma-Aldrich) at room temperature for 30 min.The reaction was terminated by incubation with 30 mM L-cysteine (Sigma-Aldrich) for 30 min at room temperature.Protein lysis was diluted to 2 M with 100 mM NH4HCO3and digested with trypsin (Hualishi Scientific, Beijing, China) at a ratio of 50:1(protein:enzyme,m/m)for 16 h at 37°C.The samples were treated with trypsin(Hualishi Scientific)at a protein:enzyme ratio of 100:1 (m/m) for 4 h.The digested lysates were desalted using tC18 cartridges (Sep-Pak, Waters, Milford, MA, USA) and dried using a SpeedVac.
TMT(Thermo Fisher Scientific Inc.,Waltham,MA,USA)reagents(6-plex or 10-plex)were used for peptides labeling according to the manufacturer's protocol.Simply,0.4 mg of TMT reagent was added to each sample and then incubated at room temperature with mild shake for 60 min.The reaction was terminated by 4 μL of hydroxylamine(5%,m/m)and mixed.The peptides were desalted by tC18 cartridges (Sep-Pak, Waters).The same control lysate (MCF7 cell lines treated with dimethyl sulfoxide (DMSO; WAK-Chemie Medical GmbH, Jena, Germany) was used across all TMT-6plex groups with two biological replicates.
The mixed samples were then re-suspended in 0.1% (V/V) trifluoroacetic acid (Sigma-Aldrich), and separated into 80 fractions using high pH reverse-phase high performance liquid chromatography (HPLC) with XBridge Peptide BEH C18column (5 μm,4.6 mm × 250 mm,130 Å, Waters).The flow rate was set at 1 mL/min.The gradient was set to 90 min from 0%to 95%of buffer(98%(V/V) acetonitrile (ACN; pH 8.5; Thermo Fisher Scientific Inc.).The 80 fractions were concatenated into 20 fractions and dried using a SpeedVac.
2.4.Liquid chromatography-mass spectrometry (LC-MS) analysis
Each fraction of the dried peptides was re-suspended in formic acid (0.1%, V/V) and separated using an EASY-nLC 1000 LC system(Thermo Fisher Scientific Inc.).The gradient was set to 70 min from 8%to 46%ACN.A home-made capillary column(75 μm i.d.×20 cm length) packed with C18beads (size 1.8 μm, 100 Å, Dikma Technologies, Beijing, China) was used.
The peptides were analyzed using an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific Inc.).The full scan range was m/z 450-1500.The maximum injection times were set to 50 and 80 ms for the full and MS/MS scans,respectively.The resolution at m/z 200 was set to 60,000 and 15,000 for the full and MS/MS scans, respectively.The automatic gain control targets was set to 500,000.Fifteen top ions were used for analysis.The high collision dissociation normalized collision energy was set to 40%.
2.5.MS data pre-processing
MaxQuant software(1.5.3.8)with human database from UniProt(96,447 sequences,downloaded on June 6,2019)was used for raw data parsing.The parameters were set with an false discovery rate(FDR) of 0.01 at the level of protein, peptide, and modification.Enzyme specificity was set to trypsin/P with tolerance of 2 missed cleavages.The fixed modification was carbamidomethyl (cysteine residues) with acetyl (protein N-term) and oxidation (M) set as variable modifications.TMT label was set to 6-plex TMT for proteomic data.The MaxQuant settings for the 10-plex labelled samples were the same with that of 6-plex labelled samples.The MaxQuant setting of 6-plex labelled samples was used for our drugresponse proteomic landscape.In addition,the MaxQuant setting of 10-plex labelled samples was used for the drug combination assays(EZH2i and topotecan).
The reverse or potential contaminant proteins were deleted.The intensities from the same gene were summed and then normalized by the median intensity for each sample.Thirty-two expression matrices were combined by gene symbols.The ratio of sample(drug treatment) abundance to control (DMSO) abundance was calculated as the relative abundance and then log2 transformed.
To balance the confidence and reservation of the scope of data set to low abundant proteins,the data set was filtered for proteins measured in 50%of all samples.The missing values was imputed by the minimum value of the data set.The batch effect was removed by the R tool,ComBat[10,11].We used time point information(P1,P2, and P3) as batch covariate while other parameters are default(par.prior = TRUE, mean.only = FALSE, BPPARAM = bpparam(“SerialParam”), mod = NULL, ref.batch = NULL) for “Combat”function.
2.6.Protein-protein interaction (PPI) networks
The PPI wasanalyzed using the STRING website(https://string-db.org/)using all of the commonly downregulated/upregulated proteins(Student's t-test,P <0.05 and fold change >1.5 in at least 60%drugs for each class type).In total,201 downregulated proteins(59 in the adenosine monophosphate (AMP)-activated protein kinase (AMPK)inhibitor,37 in the bromodomain and extraterminal(BET)inhibitor,one in the cyclin-dependent kinase (CDK) inhibitor, 22 in the DNA alkylating drug,6 in the DNA/RNA synthesis inhibitor,15 in the IMiDs,38 in the phosphatidylinositol-3-kinase (PI3K) inhibitor, 11 in the proteasome inhibitor,10 in the topoisomerase inhibitor,and 2 in the tubulin inhibitor) were identified, resulting in 160 unique proteins.Similarly,229 upregulated proteins(42 in the AMPK inhibitor,38 in the BET inhibitor, 3 in the CDK inhibitor,15 in the DNA alkylating drug, 5 in the DNA/RNA synthesis inhibitor, 2 in the histone deacetylase(HDAC)inhibitor,48 in the IMiDs,26 in the PI3K inhibitor, 28 in the proteasome inhibitor, 16 in the topoisomerase inhibitor,and 6 in the tubulin inhibitor)were identified,resulting in 187 unique proteins.The PPI networks were plotted by the R tools“igraph”.
2.7.The “querying” process
The querying signatures were obtained from the proteome data of public articles.Proteomic data were downloaded and mapped to our database using gene names.Significantly regulated genes(Student's t-test,P <0.05,fold-change >1.5)were used as querying signatures and were compared with our database using Spearman's correlation.The similarity between the querying signatures and our database was defined using the following criteria:1)the P value of Spearman's correlation should be less than 0.05; 2) the absolute correlation coefficient of Spearman's correlation should be larger than 0.2;and 3)we mainly considered the top five correlated drugs in our database.
2.8.Immunofluorescence and assessment
MCF7 cells were plated in 12-well dishes on glass coverslips and treated with colchicine (10 or 20 nM) or xanthohumol (5, 10, or 15 μM)for 24 h before staining with Tubulin-Tracker Red.Then,the cells were washed in PBS and fixed with 4% paraformaldehyde for 30 min.Next,the fixed cells were washed with 0.1%(V/V)Triton X-100 in PBS for 5 min; this was repeated twice.Subsequently, cells were incubated with Tubulin-Tracker Red for 1 h in dark and then washed thrice with 0.1% (V/V) Triton X-100 in PBS.Immunofluorescence was examined using confocal microscopy (Olympus FV1000) and photographed at ×40 or ×100.Cells were counted from six images per well (×40) with a minimum of 100 cells in total.
2.9.Determination of 20S proteasome activity
In vitro assay was performed to estimate 20S proteasome activity after treatment with different compounds.In Tris-HCl buffer(50 mM, pH 7.5), 250 ng commercial human 20S proteasome was reacted with 100 μM Suc-LLVY-AMC at 37°C for about 30 min with the treatment of 10 μM tamoxifen or 4-hydroxy tamoxifen using 0.5 μM carfilzomib as control.Hydrolyzed 7-amino-4-methylcoumarin (AMC) was measured using a microplate reader(Spark Cyto, Tecan, Zürich, Switzerland) with a filter of 360 nm excitation and 460 nm emission.
2.10.Cell viability assay
Different cell lines (CRBN+/+HCT-116 or SMMC-7721) were cultured in 96-well plates for 24 h with different compounds(including CC-885, benzethonium chloride, MLN4924, and statin drugs).After 48 h of treatment, the absorbance (450 nm) was measured for Cell Counting Kit-8 (Meilunbio) on a plate reader(Infinite 200pro,Tecan).Each assay was repeated no less than three times.The combination index (CI) was estimated using the CompuSyn (version 1.0) to define the potential synergistic effect.
2.11.Gene set enrichment analysis
Proteomic profiling data of SMCC-7721 cells treated under different conditions (DMSO, topotecan, and topotecan + GSK126)were used for gene set enrichment analysis (GSEA).The GSEA was performed using the oncogenic gene set(C6) from MsigDB(v 6.2).The permutation type was selected as “gene-set”.
2.12.Docking procedure
Docking was performed using Schr¨odinger (version 2.4, New York City,NY,USA,2010).The LigPrep module was used to prepare small molecules.The Epik was used to determine the protonation states of the molecules [12].Protein preparation and grid generation was conducted using crystal structure of DDB1-CRBN E3 ubiquitin ligase bound to lenalidomide (Protein Data Bank (PDB)ID: 4CI2) before docking.Glide module (version 6) was used to perform docking procedure with standard precision mode.The binding interactions were analyzed and displayed using PyMOL(version 1.8).
2.13.Data and materials availability
All MS raw data have been deposited to the iProX Consortium with Project ID:IPX0003183000(URL:https://www.iprox.cn/page/project.html?id=IPX0003183000).
3.Results
3.1.Construction of the dynamic proteomic landscape of drug response in MCF7 cell line
To establish a systematic study of perturbed proteomes induced by the 78 distinct bioactive compounds, we used a 6-plex TMT labelled proteomic approach with biological replicates in the MCF7 cell line(Fig.1A).An overview of these small molecules is provided in Table S1, which covers almost all types of anticancer drugs.The targets of the selected compounds were involved in a broad range of signaling pathways, tyrosine kinases, proteasomes, DNA synthesis/repair enzymes and epigenetic enzymes (Fig.1B and Table S1).In addition, the proteome profiles of IMiDs such as lenalidomide, thalidomide, and their derivatives, which target protein degradation, were collected in our dataset (Fig.1B).The concentrations of the compounds were selected based on their 10-fold half-maximal inhibitory concentration (IC50) values.For compounds with no apparent effect on cell viability (IC50≥1 μM),dosing concentrations were controlled at 10 μM for proteomic analysis(Table S1).The duration of the compounds was set to 12 h to avoid non-apparent signals or extensive cell death effects when the profiles were obtained too early or late.
A total of 32 batches of TMT-labelled samples were analyzed in our study.The resulting dataset contained quantitative proteomic data for >15,000 proteins(8,889 genes <1%false discovery rate,and≥1 unique peptide) (Table S2).The average number of proteins identified in each TMT group was approximately 6,400 (Figs.S1A and B).All the proteomic data presented a unimodal distribution(Fig.S1C) and passed the quality control process (Figs.S1A-E).Because the different time points for the experiments (P1, P2, and P3) that could result in the batch effect when data from multiple batches were integrated, we removed it by using a widely used Combat method (Figs.S1D and E) according to the previous study[13-15].An overview of proteomic profiling with different drug treatments revealed the dynamics of regulated proteins especially down-regulated proteins, as well as the highest and lowest abundant proteins identified by the median protein regulation across all compounds (Fig.1C).The Spearman's correlation coefficient of protein expression was found to be much higher in corresponding compounds with similar pharmacological mechanisms than in those with distinct pharmacological mechanism (Fig.1D).In addition,principal component analysis(PCA)was performed on the signatures of all the 78 compounds.As expected,most compounds with similar mechanisms clustered closer than the others(Fig.1E),such as cytotoxic drugs, epigenetic inhibitors, and inhibitors targeting the ubiquitin-proteasome system.Compared with these small molecules,compounds targeting the cell cycle were far from each other in the PCA analysis(Fig.1E),which may indicate a wide variation in the diverse mechanisms and biological effects of CDK inhibitors.Known IMiDs-dependent targets [16] were downregulated after treatment of IMiDs; however, some of them were not quantified which may be due to cell-type specificity(Fig.S1F).
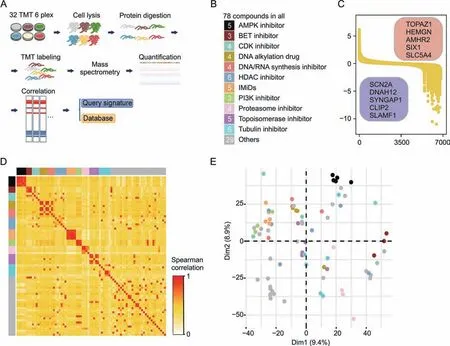
Fig.1.Proteomic landscape of drug treatment.(A) Workflow of the proteomic profiling and bioinformatic analysis.(B) Summary of the information for compounds we used.(C)Overview of the proteomic profiling with different drug treatment.Shown are the dynamics of protein abundances (log2 ratio).The highest- and lowest-abundance proteins,identified by the median of protein regulation across all compounds,are shown in the upper right and bottom-left box respectively.(D)The spearman correlation between samples with different drug treatment.The color represents different class of drugs as shown in Fig.1B.(E)The results of principal components analysis.The color represents different class of drugs as shown in Fig.1B.TMT:tandem mass tag;AMPK:adenosine monophosphate(AMP)-activated protein kinase;BET:bromodomain and extra-terminal domain;CDK:cyclin dependent kinase; HDAC: histone deacetylase; IMiDs: immunomodulatory drugs; PI3K: phosphatidylinositol-3-kinase; Dim: dimension.
3.2.The application scope of our proteomic signature resource
We further estimated the application potential of our database to understand the underlying pharmacological mechanisms using proteomic data from different cell lines,different processes of drug treatment, and different previously reported proteomic platforms.For these analyses, proteomic signatures from other groups were collected and used as input data to query the database.In this process, different therapeutic classes of drugs or bioactive “tool”compounds were included (Table S3), and the similarity was compared using proteome data in our and other groups.
A previous deep proteome dataset showed the proteomic response to treatment with nine compounds derived from different drug types in MCF7 and A549 cell lines in a TMT-based quantitated manner[4].All signatures of the nine drugs in both cell lines were used as input data for the “query” process, and detailed results of the top five drugs with high similarity (P <0.05, rho >0.2) in our dataset were obtained (Fig.2A) [4].Most of the perturbations caused by drug treatment on the proteome were recapitulated in our database in both the MCF7 and A549 cell lines.For example,only one Mdm2 inhibitor (nutlin-3a) was used to establish our database,and our data showed that its signature ranked at the top(rho = 0.45) after the query of nutlin-3a signatures in both cancer cell lines (Fig.2A).Moreover, notably higher similarity was observed between the signature of bortezomib and that of our three proteasome inhibitors (bortezomib, rho = 0.42; carfilzomib,rho = 0.4; and MG-132, rho = 0.4) in the database (Fig.2A).In addition, the signatures of the tubulin inhibitors vincristine(rho = 0.6) and colchicine (rho = 0.58) in our database were captured when the proteomic signature of vincristine treatment was used as input data (Fig.2A).A relatively low similarity was found for cytotoxic drugs, which may be due to their similar pharmacological effects after treatment with DNA synthesis inhibitors, DNA alkylating drugs, and topoisomerase inhibitors(Fig.2A).No drug in our database was similar to raltitrexed(a DNA synthesis inhibitor).For querying the proteomic signature of two tyrosine kinase inhibitors (dasatinib and gefitinib), we only found that the profiles of the BCR/ABL kinase inhibitor imatinib or Src/Abl inhibitor bosutinib were in the top five ranks but other tyrosine kinase inhibitors were out of the top five ranks (Fig.2A), which indicated that the signature, at the phosphorylation level but not the protein level, may be more representative of kinase inhibitors.Our results indicate that our database can be utilized for the MCF7 cell line as well as other cell lines.
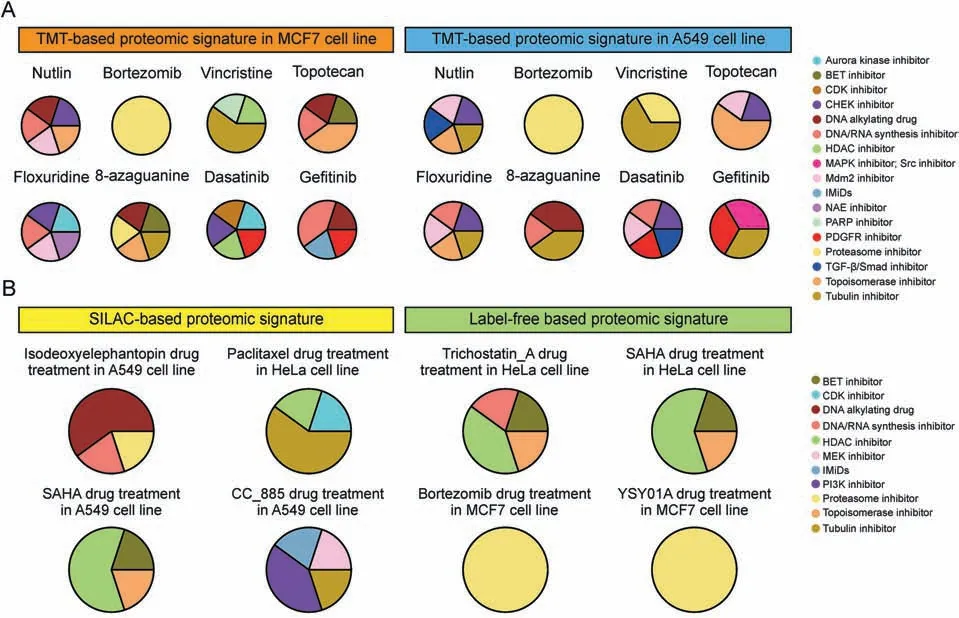
Fig.2.Proteomic alteration revealed the mechanism of drug effect.(A) The querying results of our database using signatures from published proteome data [4] based on Tandem Mass Tag(TMT)labeling method.(B)The querying results of our database using signatures from published proteome data[17-22]based on stable isotope labeling by amino acids in cell culture(SILAC) labeling and label-free method.BET:bromodomain and extra-terminal domain; CDK:cyclin dependent kinase; CHEK: checkpoint;HDAC:histone deacetylase;MAPK:mitogen-activated protein kinase;Mdm2:murine double minute 2;IMiDs:immunomodulatory drugs;NAE:NEDD8-activating enzyme;PARP:poly(adenosine diphosphateribose)polymerase;PDGFR:Platelet-derived growth factor receptor;TGF:transforming growth factor;SAHA:suberoylanilide hydroxamic acid;PI3K:phosphatidylinositol-3-kinase.
We next explored whether our TMT-based drug signature database could also be used to explore pharmacological mechanisms using proteomic signatures produced from other quantification methods, such as Stable isotope labeling by amino acids in cell culture (SILAC) or label-free quantitative proteomic methods.Our results showed that the proteomic signatures from different types of drugs acquired using both SILAC and label-free proteomic strategies [17-22] correlated well with the proteomic profiles of similar drugs identified in our database (Fig.2B).
These results showed the broad application potential of our dataset in exploring pharmacological mechanisms using the proteomic data produced by our group and others, although the differences were unavoidable in fields such as cancer cell types,proteomic strategies(TMT-based,SILAC-based,and label-free-based methods),compound concentrations, and duration.This result is consistent with those obtained at the transcriptomic level [1].However, we could not ignore the“false positive”or“false negative”query results,which may be due to the initial experimental designs of our and other groups.
3.3.The characterization of commonly changed proteins under the treatment of different drugs
Next, to explore the potential molecular mechanism of drug activity, we determined the commonly regulated proteins in each drug type(Student's t-test,P <0.05 and fold change >1.5,in at least 60%of drugs for each drug type).Using this criterion,we identified 187 commonly upregulated proteins and 160 commonly downregulated proteins.To explore the correlation between each type of drug perturbation,we constructed PPI networks for these proteins using the String database (Fig.3A), in which 78 upregulated proteins with 128 interactions and 94 downregulated proteins with 432 interactions were acquired (Fig.3A).Interestingly, more interactions were observed among commonly downregulated proteins than among commonly upregulated proteins.Interactions among commonly regulated proteins may indicate complementary possibilities of different drug perturbations.
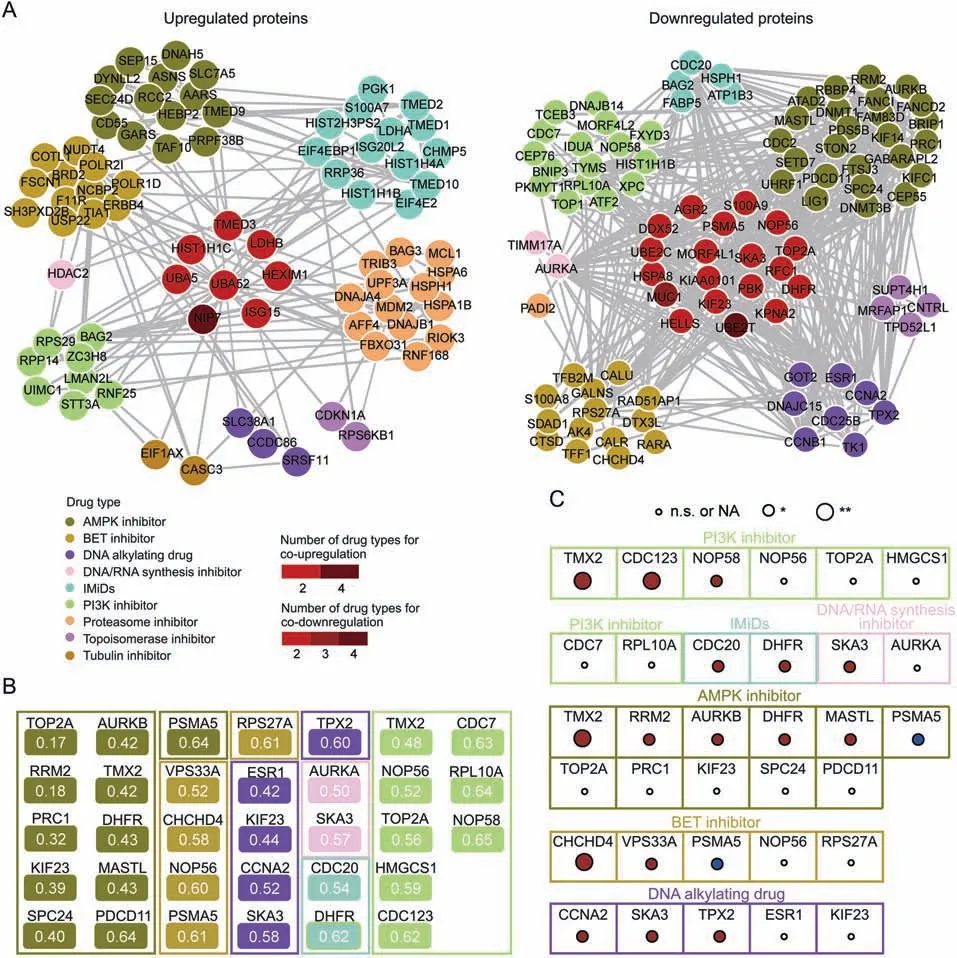
Fig.3.Proteomic alteration by the treatment of each drug type.(A) Protein-protein interaction (PPI) networks of upregulated/downregulated proteins from nine class of compounds.(B) The representative downregulated proteins with the CRISPR-Cas9 gene knockout scores (CERES) <-0.6 of MCF7 cell line and the mean CERES <-0.6 of 12 estrogen receptor (ER) positive breast cancer cell lines for each type of drugs.The number shows mean of fold-change for protein in each type of drugs.(C) The prognosis power of the potential key proteins.The color of circle represents the protein expression positively(red)or negatively(blue)correlated with the poor prognosis in breast cancer.Log-rank P-value was used for the significance.*P <0.05 and **P <0.01;n.s.:no significance.NA:not available.AMPK:adenosine monophosphate(AMP)-activated protein kinase;BET:bromodomain and extra-terminal domain; IMiDs: immunomodulatory drugs; PI3K: phosphatidylinositol-3-kinase.
To identify more important proteins for the commonly downregulated proteins of each type of drug perturbations,we estimated the gene dependence of all the commonly downregulated proteins in each drug class using the DepMap database(https://depmap.org/portal/download/).The average gene essentiality scores (CRISPRCas9 gene knockout scores (CERES)), which represent gene dependence, were calculated for the 12 estrogen receptor (ER)-positive breast cancer cell lines (similar to MCF7 in genetic background; Table S4) for the downregulated proteins (Fig.3B).The results showed downregulated proteins with both CERES <-0.6 in MCF7 cell line and average CERES <-0.6(defined as essential genes[23]) in 12 ER-positive breast cancer cell lines as the potential key proteins for pharmacologic activity(Fig.3B).Interestingly,four U.S.Food and Drug Administration (FDA)-approved drug targets defined in the Human Protein Atlas (HPA) database (https://www.proteinatlas.org/search/protein_class:FDA+approved+drug+targets) were identified as potential key proteins for pharmacological activities, including TOP2A, RRM2, DHFR, and ESR1.Furthermore,we explored the prognostic power of the above potential key proteins using The Cancer Genome Atlas breast cancer data analyzed in the HPA database (https://www.proteinatlas.org/humanproteome/pathology).Our results also indicated that for each drug type,higher expression of some key proteins was positively associated with poor prognosis (Fig.3C).The co-inhibition of these key proteins may have positive effects on breast cancer.We provided resources for the discovery of new therapies.
3.4.Our dataset revealed pharmacological mechanism of natural products
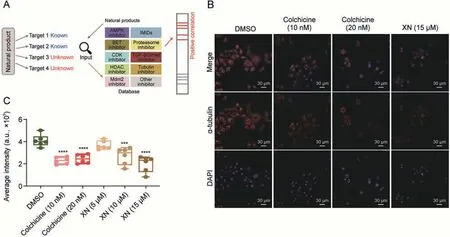
Fig.4.Drug mechanism querying of natural products.(A)Workflow of the drug mechanism prediction of natural product.(B)MCF7 cells treated with dimethyl sulfoxide(DMSO),colchicine, or xanthohumol (XN) for 24 h.Then the α-tubulin morphology was detected by immunofluorescence with anti-α-tubulin and 4',6-diamidino-2-phenylindole (DAPI)staining.Shown was the representative images which visualize alpha-tubulin (red) and nuclear (blue).(C) Average immunofluorescence intensity was count from 6 different regions.One-way analysis of variance was used in the analysis.Compared with DMSO, ***P <0.001 and ****P <0.0001.AMPK:adenosine monophosphate(AMP)-activated protein kinase; IMiDs: immunomodulatory drugs; BET: bromodomain and extra-terminal domain; CDK: cyclin dependent kinase; TGF: transforming growth factor; HDAC: histone deacetylase; Mdm2: murine double minute 2.
Natural products, originating mainly from plants and microorganisms, have diverse chemical skeletons with broad biological activities [24].Nearly 40% of FDA-approved compounds were natural products in the past 30 years[24-28].Because natural products are not rationally designed based on known targets,they have been reported to have multiple targets.Our dataset then provided an opportunity for exploring new mechanism based on the “query assay” of proteomic signatures of natural products (Fig.4A).The proteomic data with statistical significance (Student's t-test,P <0.05,fold-change >1.5)from previous study[29-32]were used as signatures (Table S5) queried in our database, and drugs with similar profiles tended to appear at the top of the list with a highly positive Spearman's correlation(P <0.05,rho >0.2)(Fig.4A).
We queried the proteomic signature of Rosemary extract from a previous study in our database [29] (Table S5).Our results showed that the three different proteasome inhibitors presented a high positive correlation (rho = 0.54 for carfilzomib, rho = 0.52 for MG132,and rho=0.45 for bortezomib),indicating that some major components in Rosemary may inhibit proteasome activity.This hypothesis was ultimately confirmed by a previous study showing that carnosol, a dietary diterpene and major polyphenol in Rosemary extract,inhibited 20S proteasome proteolytic activity[33].Similarly,signature of extract of Lippia origanoides[30](Table S5)was found to be similar to proteasome inhibitors in function (rho = 0.24 for MG132, rho = 0.22 for carfilzomib), which was confirmed by a previous study, as the polyphenol quercetin and apigenin were reported to be abundant in it and showed inhibition of the 20S proteasome[34].In addition,the cyclin dependent kinase inhibitor Ro-3306 ranked at the top of the list after the signature query of odoroside A (rho = 0.27), and its potential activity in causing cell cycle arrest was supported by previous studies[31].
Chalcones exhibit anticancer activities, induce cell apoptosis,disrupt angiogenesis,and inhibit tubulin assembly[32].We queried the proteomic signature of xanthohumol (a prenylated chalcone found in hops)[32]in our database(Table S5).The results showed that its proteomic pattern was similar to that of the tubulin inhibitors colchicine (rho = 0.39) and vincristine (rho = 0.37).Thus,we explored whether xanthohumol inhibited tubulin assembly using an immunofluorescence assay (Fig.4B).Our results clearly showed that the fluorescence shape size of the cytoskeleton was relatively decreased in a dose-dependent manner when cells were treated with xanthohumol (Fig.S2).The results also showed decreased fluorescence intensity with an increased concentration of xanthohumol in MCF7 cells (one-way analysis of variance,P <0.05),with the tubulin inhibitor colchicine as a positive control(Fig.4C).This result indicates the predictive power of our database and suggests a new strategy for its application in natural product discovery and development.
3.5.Proteomic signatures from clinically approved drugs identified“off-target” pharmacological mechanisms
The polypharmacology of drugs presents multiple-target activity,leading to adverse effects on human safety.Meantime, the unprecedented efficacy of polypharmacological drugs for the treatment of multigenic diseases presents opportunities for modern drug discovery and drug repurposing[35].Therefore,effective inspection of unknown mechanisms for FDA-approved drugs could not only compromise drug safety, but also confer superior therapeutic efficacy.By comparing the proteomic perturbation profiles of clinical drugs with the signatures derived from our database, we could predict potential “off-target” pharmacological effects based on the positive Spearman's correlation result(P <0.05,rho >0.2)(Fig.5A).

Fig.5.Off-target effect prediction of U.S.Food and Drug Administration(FDA)-approved drugs.(A)Workflow of the off-target effect prediction using published proteome profiling treated with US FDA-approved drug.(B) The in-vitro validation of the 20S proteasome inhibition.The prediction used signatures from the published proteome data [37].(C) The pharmacological activities of CC-885(25 nM)(left)and statin drugs(right)in HCT116 cell line.One-way analysis of variance was used in the analysis(n=6 for each group).(D)Cell viability of MCF7 or cereblon(CRBN)knockout(KO)MCF7 cells treated with lovastatin(LOV,25 μM)at different dose.Student's t-test was used in the analysis(n=6 for each group).(E) Molecular docking result of LOV with CRBN protein.*P <0.05, **P <0.01, ***P <0.001, and ****P <0.0001.DMSO: dimethyl sulfoxide; MLN: MLN4924; WT: wide type.
Tamoxifen and 4-hydroxytamoxifen, which are selective estrogen receptor modulators,have been used clinically to treat patients with ER-positive breast cancer [36].We extracted the proteomic signature of 4-hydroxytamoxifen(Table S5)in a previous study[37]and used this signature for database “query” process.Surprisingly,we found that the proteomic fingerprints of 4-hydroxytamoxifen presented potentially diverse pharmacological activities, such as an AMPK activator (metformin), a proteasome inhibitor(carfilzomib), an Mdm2 inhibitor (Nutlin-3a), and an epigenetic inhibitor (valproic acid and JQ-1).A previous study reported that the tamoxifen derivative ridaifen-F(Fig.S3A)directly inhibited the human 20S proteasome [38].Therefore, we tested whether tamoxifen and 4-hydroxytamoxifen could function as potential proteasome inhibitors.Our results clearly showed that tamoxifen and 4-hydroxytamoxifen could inhibit chymotrypsin-like activity in 20S purified proteasome with Suc-LLVY-AMC labelled peptide as a substrate and carfilzomib as a positive control (Fig.5B).These results indicate the potential application of our database for exploring pharmacological effects.
Using the same “query” strategy, we found lovastatin [39] and benzethonium chloride [40] (BZN) may have similar pharmacological mechanism with IMiDs (Table S5).IMiDs such as thalidomide and its analogs can bind to the cereblon(CRBN)subunit of the CRL4 CRBN E3 ubiquitin ligase, which confers ubiquitination and proteasomal degradation of specific substrates.This pharmacological mechanism has been clinically approved for the treatment of multiple myeloma [41,42].In addition, neddylation inhibitors such as MLN4924 can inhibit the activity of cullin-RING E3 ligases and reverse the anticancer activity of thalidomide and its derivatives [43].We then conducted a cell viability assay using lovastatin and its derivative atorvastatin in a MLN4924-dependent manner with CC-885 (a novel thalidomide derivative with IMiDs activity) as a positive control.The results showed that lovastatin inhibited the cellular survival rate, and the addition of MLN4924 prevented the cytotoxic activity of these two statin drugs, which was consistent with the pharmacological results for CC-885(Fig.5C).In addition, two other statin drugs (atorvastatin and revastatin) showed similar pharmacological activities (Fig.5C),indicating their potential “off-target” effects.The combination of benzethonium chloride and MLN4924 did not have a similar effect(Fig.S3B).Next, we compared the viability of CRBN+/+and the CRBN-/-cell lines treated with lovastatin to explore the relationship of statin drugs with CRBN subunit,and the results showed that significantly stronger cytotoxic activity of lovastatin was observed in CRBN+/+cell line in increasing drug dose(Fig.5D).These results indicated a possible dependency of CRBN on the pharmacological activity of lovastatin.This hypothesis was further supported by molecular docking results, which showed a potential interaction between lovastatin and the CRBN subunit (Fig.5E).
Polypharmacology of small molecule was mostly related to their“off-target”effects and was a puzzling question in their clinical use or rational design of synergistic combinations [44-46].Our proteomic pattern-based study contributes to a key element in drug discovery in view of this problem.
3.6.Rational design of synergistic drug combinations by the complementary proteomic profiles

Fig.6.Combination therapy prediction for drug resistant exploration.(A)Workflow of the combination therapy prediction using published proteome profiling treated with drugs in resistant versus sensitive cells.(B)The combination index for topotecan+GSK126 combination at different dose in SMMC-7721 cells.The prediction used signatures from published proteome data[48].(C) The oncogenic pathway analysis for proteomic profiling of SMMC-7721 cells treated with dimethyl sulfoxide (DMSO), topotecan,and topotecan+GSK126 combination.(D)The cell viability of SMMC-7721 cells treated with different drugs for triple-combination therapy validation.One-way analysis of variance was used in the analysis.*P <0.05 and ****P <0.0001.GSK126:10 μM;topotecan:500 nM;AZD:AZD8055(50 nM).AMPK:adenosine monophosphate(AMP)-activated protein kinase;NF-κB:nuclear factor kappaB; CDK: cyclin dependent kinase; CHEK: checkpoint; HDAC: histone deacetylase; JAK: janus kinase; NA: missing value; FDR: false discovery rate.
Developing highly selective molecules targeting a definite“driver”molecular event was as exciting as“targeted therapy”,but its major limitation was the drug resistance through rewiring of cellular signaling network[47].Our proteomic platform can also be used to elucidate mechanistic biomarker candidates and identify drug combination strategies(Fig.6A).For primary drug resistance,the proteomic profiles of sensitive and resistant cell lines can be acquired with or without treatment with the corresponding drugs.The difference between the proteomic signature under drug stimulation could be used as input data for “query” process, and compounds with negative Spearman's correlation coefficient(P <0.05,rho <-0.2) may be combined drugs utilized in the resistant cell line.Similarly, for acquired drug resistance, the proteomic profile difference between the sensitive and acquired resistant cell lines was utilized as input data, and drugs in our database were considered as potential combined drugs when the corresponding Spearman's correlation coefficients were negative (P <0.05, rho<-0.2) (Fig.6A).We then used our dataset to explore potential drug combinations.
Our previous study reported one of the key drug resistance mechanisms of the methyltransferase EZH2 inhibitor (EZH2i) in solid tumors and showed that combined inhibition with BRD4 could increase the sensitivity of EZH2i in solid tumors[48].To validate the power of our database in exploring potential drug combination,we queried the proteomic signature of EZH2i in primary resistant and sensitive cell lines acquired previously [48] (Table S6).Our results showed that the BRD4 inhibitor, JQ1, had a negative Spearman's correlation coefficient (rho = -0.23) with EZH2i resistance.This drug combination of EZH2i and JQ1 was then validated in an EZH2i resistant cell line in our previous study, which indicated the utilization potential of our database for predicting drug combination[48].In addition, we determined that the topoisomerase inhibitor topotecan presented a notably higher negative Spearman's correlation coefficient(rho=-0.46),suggesting its synergistic effect with the EZH2 inhibitor.Cell viability was significantly different between the individual drugs and the combination of the two drugs(Fig.S4A).We then calculated the CI of EZH2i and topotecan in EZH2i resistant SMMC-7721 cell line, and the results showed a synergistic effect (combination index <1) between the EZH2 inhibitor GSK126 and topotecan at different doses (Fig.6B and Table S6).To determine the molecular mechanism of this synergistic effect, we conducted a TMT-based proteomic strategy to compare the dynamic proteome changes in topotecan treatment with a combination of the two drugs, using DMSO as a control in three biological replicates (Fig.S4B).The GSEA pathway analysis (C6:oncogenic pathway) showed several oncogenic pathways were downregulated, including VEGF, TGFB, MEK, etc., by the combined inhibition of EZH2 and topoisomerase compared with the single treatment with topotecan and DMSO (Fig.6C).It may be the potential reason for the synergistic effect of these two drugs.However,we also found that several oncogenic pathways were upregulated in the combination group, in which the mammalian target of rapamycin (mTOR) signaling kinase pathway was highly enriched(Fig.6C).This result suggests that the triple-combination inhibition of EZH2,topoisomerase, and mTOR signaling pathway may display better efficacy, which was further proven by cell viability experiments with the treatment of these three drugs (Fig.6D).
Next,we explored potential combination strategies for acquired drug resistance by querying the proteomic differences between acquired resistant and sensitive cell lines[49-54](Table S6).Based on the negative correlation,corresponding drug combinations were estimated to potentially overcome different types of drug resistance (Table S6).
Therefore, a system-wide proteomic approach at the protein level integrated with pharmacological analysis could raise rational design of potential drug combinations and facilitate new therapeutic options.
4.Discussion
Studying intracellular dynamic molecular networks perturbed by drug stimulation can provide useful information for revealing MoAs and “off-target” effects of compounds.Perturbation experiments on cancer cell lines have advanced our understanding of the biological consequences of phenotypic changes under stress [55].Recently, pharmacologic and genomic perturbation studies have been conducted on cell lines at the genomic level, in which genome-wide “cancer dependency maps” [23,56,57] and transcriptome-wide “Connectivity Map” [58] have pioneered this concept and provided a system-level view of phenotypic and cellular effects in cancers.Large-scale drug signature profiles at the transcriptional level have been successfully utilized to identify connections between different compounds, characterize novel pharmacological mechanisms, and explore new drug uses [1,58].However, the relatively low correlation coefficient between transcriptomic and proteomic data,as reported in both cell lines[59,60]and clinical tumor samples [61], reveals the limitations of using mRNA data as a reliable readout.Proteomes, being directly influenced by chemical substances, exhibit superior specificity for compound interference compared to transcriptomes.Therefore,reverse-phase protein array or MS-based protein-level readout assays have been used to quantitatively predict how cancer cells employ their adaptive protein response under the stress of drug treatment [4-7].These two proteome-wide methods are complementary due to their identification scales and study purposes,which highlight the value of the systematic proteomic characterization of behaviors or adaptations in cancers.
In this study, we present a TMT-based proteomic platform for the global proteome perturbation profiling of 78 compounds in the MCF7 cell line, which covers almost all types of anticancer drugs.We explored the functional relevance of our dataset using a“querying”process based on the similarity of proteomic signatures acquired in other studies.We uncovered the microtubuleinhibitory activity of the chalcone xanthohumol, and the potential“off-target” effects of tamoxifen and lovastatin.In addition, the protein responses of different compounds were used for the rational design of drug combination therapies, and we found a synergistic effect between the EZH2 inhibitor GSK126 and the DNA synthesis inhibitor topotecan.These results indicate the successful utility of our protein response dataset,which facilitates therapeutic opportunities for new uses of conventional drugs.
We also recognize the current limitations of the rational design of our project.First,we controlled concentration at 10 μM for most of the compounds we used, and the duration was set as 12 h.However, this design is somewhat unsuitable for compounds such as epigenetic inhibitors.Second,we only used the MCF7 cell line in our database.As an important cell line for drug screening,MCF7 has been used to identify transcriptomic drug responses [1].Although our dataset was suitable for query processing of proteome signatures acquired in other cancer cell lines, some false negative or positive results were unavoidable,such as the BZN signature(from the A549 cell line).This may have resulted from the difference in the origin of the tumor tissues, as we only used the MCF7 cell line for dataset establishment.Mutation information(Dep-Map Public 19Q3[62], https://depmap.org/portal/download/), such as PIK3CA E545K and GATA3 frameshift mutations, may contribute to proteomic signatures.We also found a significant difference (Fisher's exact test,P = 0.0073) in the number of non-silent mutations between the MCF7 and A549 cell lines.The power(n=2 for each drug treatment)was unsuitable for multiple testing.Third, proteomic signatures at the post-translational modifications(PTM)level are also important,in which dynamic PTM changes in phosphorylation, epigenetic modification, acetylation and ubiquitination directly reflect molecular perturbations in signaling pathways, gene expression, energy metabolism, and protein turnover events, respectively.Therefore,the integration analysis of proteomic and diverse PTM-omics with phenotypic drug response or pharmacological data provides new insights into drug combinations, especially for kinase inhibitors,ubiquitination system modulators, epigenetic inhibitors, and metabolism inhibitors.Additionally, we noted that the first hit of some signatures was not the drug type itself, such as for 8-azagunaine, and no drug in our database was similar to raltitrexed(a DNA synthesis inhibitor).This discrepancy may be due to the loss of drugs with a signature similar to that of 8-azaguanine and raltitrexed,indicating the incompleteness of our database.
5.Conclusions
In conclusion, our data portal raised functional connectivity by comparing proteome profiles to identify novel biological effects,which promoted the integrated analysis of various data types and facilitated the investigation of drug behaviors in cancer therapy.
CRediT author statement
Zhiwei Liu:Data curation,Formal analysis,Investigation,Methodology, Visualization, Writing - Original draft preparation,Reviewing and Editing;Shangwen Jiang: Investigation, Methodology;Bingbing Hao,Shuyu Xie, andYingluo Liu: Validation;Yuqi HuangandHeng Xu:Formal analysis;Cheng Luo:Writing-Original draft preparation;Min HuangandMinjia Tan:Writing-Reviewing and Editing;Jun-Yu Xu: Conceptualization, Investigation, Methodology,Writing-Original draft preparation,Reviewing and Editing.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the Natural Science Foundation of China(Grant Nos.:22225702 and 32322048),the National Key R&D Program of China (Grant No.: 2020YFE0202200), the Shanghai Academic/Technology Research Leader Program, China(Grant No.:22XD1420900), Guangdong High-level New R&D Institute, China(Grant No.:2019B090904008), Guangdong High-level Innovative Research Institute, China (Grant No.:2021B0909050003), the Shanghai Rising-Star Program,China(Grant No.:22QA1411100),the Youth Innovation Promotion Association of the Chinese Academy of Sciences (Grant No.: 2021276), the Young Elite Scientists Sponsorship Program by China Association for Science and Technology,China (Grant No.: 2022QNRC001), and the open fund of State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University,China (Grant No.: KF-202201).We also thank the support of the Innovative Research Team of High-Level Local Universities in Shanghai, China (Grant No.: SHSMU-ZDCX20212700) and Sanofi scholarship program.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.08.021.
 Journal of Pharmaceutical Analysis2024年1期
Journal of Pharmaceutical Analysis2024年1期
- Journal of Pharmaceutical Analysis的其它文章
- Lipid metabolism analysis in esophageal cancer and associated drug discovery
- Push forward LC-MS-based therapeutic drug monitoring and pharmacometabolomics for anti-tuberculosis precision dosing and comprehensive clinical management
- The role of innate immunity in diabetic nephropathy and their therapeutic consequences
- Metformin: A promising clinical therapeutical approach for BPH treatment via inhibiting dysregulated steroid hormones-induced prostatic epithelial cells proliferation
- Epimedin B exhibits pigmentation by increasing tyrosinase family proteins expression, activity, and stability
- Hydralazine represses Fpn ubiquitination to rescue injured neurons via competitive binding to UBA52