
负载碳酸钙的水包油包固乳液稳定性及相互作用研究
2024-03-20 14:51李功伟张杰郝佳赵伊聪许朵霞曹雁平
食品与发酵工业 2024年5期
李功伟,张杰,郝佳,赵伊聪,许朵霞,曹雁平
(北京工商大学 食品与健康学院,北京市食品添加剂工程技术研究中心,北京,100048)
钙是人体内最丰富的矿物质元素,对于维持人体生理平衡及健康起着关键作用[1]。全球平均膳食钙摄入量分布显示许多国家膳食钙摄入量都相对较低[2]。碳酸钙(CaCO3)具有成本低、钙含量高的优势,是目前国内外常用的强化钙[3]。因此,在食品领域,乳粉以添加碳酸钙为主,且市面上补钙产品多为碳酸钙。然而,碳酸钙作为一种难溶性钙盐在食品体系中存在分散稳定性差,贮藏过程中易析出的问题。因此,如何提高碳酸钙在液态食品中的分散稳定性受到了国内外学者的广泛关注[4-5]。
水包油包固(solid-in-oil-in-water,S/O/W)三相载体乳液作为食品营养素的载体,具有制备成本低、工艺简单等优点。目前国内外基于S/O/W技术对酶制剂、益生菌载体构建方面已有报道,但对营养素的保护研究较少。ZHANG等[6]将乳糖酶喷雾干燥形成粉末,基于S/O/W技术形成微球提高乳糖酶活性,乳糖酶稳定性较传统乳状液得到显著提高;WEI等[7]研究负载咖啡酸苯乙酯的S/O/W乳液,有效地将咖啡酸苯乙酯递送至结肠,并实现12 h的延迟释放,提高了其生物利用度和活性。
黄原胶(xanthan gum, XG)是一种阴离子胞外多糖,能够与水分子结合形成氢键从而起到增稠效果,因此可用于增加乳液黏度来提高乳液稳定性[8]。海藻酸丙二醇酯(propylene glycol alginate, PGA)是一种两亲性阴离子多糖,分子链中的糖醛酸具有亲水性,丙二醇基团具有亲油性,有较好的界面活性[9]。因此,将PGA与XG复凝聚可改善乳液的界面活性和流变特性,从而提高S/O/W乳液的稳定性。
本文以PGA和XG复合物为基质,研究二者复配界面活性及相互作用方式。基于S/O/W技术构建负载碳酸钙的S/O/W钙-脂质乳液,系统分析乳液稳定性、微观结构、流变特性。研究结果可丰富构建S/O/W传递系统理论,为开发新型营养素输送载体提供理论和技术基础。
1 材料与方法
1.1 材料与试剂
轻质碳酸钙,郑州双腾实业有限公司;精制猪油,临沂新程金锣肉制品集团有限公司;PGA(纯度大于98%),上海源叶生物科技有限公司;XG(纯度大于99.5%),北京索莱宝科技有限公司;氟罗里硅土,上海麦克林生化科技股份有限公司;尼罗红、卡尔科弗卢尔荧光增白剂,Sigma-Aldrich(上海)贸易有限公司。
1.2 仪器与设备
Smart-N超纯水机,力新仪器(上海)有限公司;Eutech pH 700 测量仪,美国Eutech公司;TB-214分析天平、TB-1102电子天平,美国Denver Instrument公司;DF-101S集热式恒温加热磁力搅拌器,河南省予华仪器有限公司;H/T16MM高速常温离心机,湖南赫西仪器装备有限公司;T-25 digital高速分散机,艾卡(广州)仪器设备有限公司;Attension Theta Flex 光学接触角仪,瑞典百欧林科技有限公司;LUMiSizer稳定性分析仪,罗姆(江苏)仪器有限公司;Zetasizer Nano ZS90纳米粒度电位仪,上海思百吉仪器系统有限公司;Mars iQ Air HAAKE哈克流变仪,赛默飞世尔科技(中国)有限公司;尼康AX共聚焦显微镜,尼康仪器(上海)有限公司;Microtrac S3500激光粒度仪,美国Microtrac公司;SU8020场发射扫描电镜,日本Hitachi公司;iS10 FTIR光谱仪,美国Nicolet公司;D8 Advance X射线衍射仪,德国Bruker公司。
1.3 实验方法
1.3.1 PGA和XG溶液的制备
分别称取一定质量的PGA粉末、XG粉末与磷酸盐缓冲溶液(PBS)混合,在50 ℃水浴搅拌2 h,之后在4 ℃过夜水合,确保其充分溶解,获得质量分数为0.6%的PGA溶液和XG溶液。
将制备得到的PGA溶液和XG溶液按0∶6、1∶6、2∶6、3∶6、4∶6、5∶6、6∶6(质量比)的比例复配,在50 ℃磁力搅拌2 h,获得总碳水化合物质量分数为0.6%不同PGA-XG配比的溶液。
1.3.2 精制猪油的纯化
实验采用精制猪油作为油相。由于市售猪油存在少量抗氧化剂和其他杂质,在实验前我们对其进行纯化以避免对实验结果造成影响。参考GAONKAR[10]的方法并稍作修改,纯化步骤如下:将氟罗里硅土按照3%(质量分数)的比例添加到融化的猪油中,70 ℃水浴搅拌2 h后,10 000 r/min离心20 min去除沉淀。随后重新加入氟罗里硅土反复操作,直至20 min内PBS在纯化猪油界面的表面张力没有明显变化。纯化后油相的密度为0.833 3 g/cm3,PBS在油水界面的表面张力为(36.79±0.77) mN/m。
1.3.3 油水界面表面张力的测量
采用悬滴法测量油水界面的表面张力[11]。将不同PGA-XG配比的溶液加入外径为2.08 mm的注射器中并排出气泡,然后将针头插入装有纯化猪油的矩形石英比色皿。随后,每个样品挤出12 μL至针尖并用自动进样装置将样品压成完整的滴状。摄像机系统立即连续捕捉液滴轮廓的变化,使用杨-拉普拉斯方程实时计算不同时间点样品的表面张力,整个过程持续20 min。实验过程中尽量避免外部振动的干扰,所有样品重复3次。
1.3.4 S/O/W钙-脂质乳液制备
S/O/W钙-脂质乳液的制备参考LI等[12]的方法并稍作修改。将食品级碳酸钙与70 ℃加热融化后的猪油按1∶10(质量比)的比例混合,在30 ℃磁力搅拌1 h,获得均一的S/O相。随后,分别移取5%(质量分数)S/O相加入1.3.1节制备的不同PGA-XG配比的W相溶液,15 000 r/min下高速剪切5 min,获得S/O/W钙-脂质乳液。
1.3.5 S/O/W钙-脂质乳液稳定性分析
采用LUMiSizer稳定性分析仪,通过STEP-Technology快速测定乳液的稳定性。不稳定性指数(instability index)是在离心的过程中,近红外光照射在样本试管上,通过记录样品不同位置的透射率分布的变化,而确定乳液中颗粒的迁移过程。实验过程中,取乳液约0.4 mL,均匀注射至样品试管底部,温度设定为25 ℃,离心转速4 000 r/min,样品的透射率的特征线每60 s记录一次,共255次。
1.3.6 S/O/W钙-脂质乳液Zeta-电位测量
用PBS将新鲜制备的乳液稀释400倍,于25 ℃下进行Zeta-电位的测定。平衡时间120 s,以3次测量结果的平均值来表示。
1.3.7 S/O/W钙-脂质乳液表观黏度测量
采用CC27型号转子在25 ℃,剪切速率0.1~200 s-1条件下对乳液进行剪切测试,获得不同剪切速率下乳液的表观黏度。
1.3.8 S/O/W钙-脂质乳液激光共聚焦显微镜观察
乳液染色参考DU LE等[13]的方法并稍作修改。取1 mL新鲜制备的乳液加到10 mL离心管,随后加入200 μL卡尔科弗卢尔荧光增白剂(激发波长405 nm)和20 μL尼罗红[质量浓度为1 g/L的二甲基亚砜溶液,激发波长488 nm]染料,旋涡10 s,平衡1 h,分别对PGA-XG复合物和油脂染色。吸取10 μL染色乳液滴加至载玻片,缓慢盖上盖玻片,避免产生气泡,随后使用60倍油镜对乳液进行观察,获取其微观结构。
1.3.9 S/O/W钙-脂质乳液粒径分布测量
使用S3500激光粒度仪在25 ℃下对乳液粒径分布进行测量,样品以水(折射率1.33)为介质输送至测量单元,重复3次。
1.3.10 PGA-XG复凝聚结构表征
1.3.10.1 激光共聚焦显微镜(confocal laser scanning microscope,CLSM)
采用1.3.8节的方法对不同PGA-XG配比的溶液进行染色,并进行激光共聚焦显微镜观察。
1.3.10.2 扫描电镜形貌观察
将单一的PGA溶液、XG溶液和PGA-XG复凝聚溶液冻干,随后使用场发射扫描电镜观察样品的表观形貌。
1.3.10.3 傅里叶变换红外光谱测量
采用红外光谱仪对冷冻干燥后的样品进行红外光谱测量。波数范围400~4 000 cm-1,信躁比是50 000∶1, 扫描32次,记录各样品的红外光谱。
1.3.10.4 X射线衍射分析
采用X射线衍射仪对冷冻干燥后的样品进行X射线衍射分析。管电流40 mA,管电压40 kV,光谱范围5°~50°,扫描速率2°/min,记录各样品的X射线衍射图谱。
1.4 数据统计与分析
实验所有样品测量都至少重复3次,结果以测量值的平均值±标准差表示。采用SPSS 17.0软件对数据进行单因素方差分析(ANOVA)和邓肯检验(P<0.05),使用Origin 2022b软件绘制图表。
2 结果与分析
2.1 不同PGA/XG复配比在油水界面的表面张力分析
油相和水相界面层的稳定性是影响乳液稳定性的一个重要因素,油水界面表面张力越小,界面层则越稳定[14]。因此,比较了单一XG和不同PGA/XG复配比在油水界面的表面张力。如图1所示,纯化猪油中PBS的表面张力为(36.79±0.77) mN/m。XG在纯化猪油中的表面张力为20.36 mN/m,这是因为XG的疏水键较少,界面活性相对较低。然而,PGA和XG复配之后大幅降低了油水界面的表面张力,这是因为PGA是两亲性化合物,其分子中的丙二醇基团可以与油脂结合,糖醛酸具有的亲水末端含有大量羟基和部分羧基,可与XG结合[15]。ZHU等[16]研究表明氢键或疏水相互作用对界面张力的降低可能是油相和水相形成稳定界面层的原因。因此,PGA和XG复合物有助于维持油相和水相界面层的稳定性。

图1 不同PGA/XG复配比在油水界面的表面张力Fig.1 Interfacial tension of PGA/XG complex with different ratios at the oil-water interface
2.2 不同PGA/XG复配比制备S/O/W钙-脂质乳液稳定性
图2-a显示了测试过程中乳液样品不同位置的原始透射曲线,从图中可以看出单一XG、PGA-XG(5∶6)和PGA-XG(6∶6)3个样品有轻微碳酸钙沉淀以及乳析现象的发生。根据透射率的变化计算得到乳液样品的不稳定指数如图2-b所示。根据不稳定指数可将样品区分为3个区域,单一XG、PGA-XG(1~4∶6)和PGA-XG(5∶6和6∶6)。随着PGA比例的增加,乳液不稳定指数先减小后增加,在PGA-XG(4∶6)之后发生徒长,第一区域至第二区域变化的原因可能是由于单一XG在油水界面表面张力较大,表面活性较低,PGA加入后改善了油水界面层的稳定性。第二区域至第三区域变化的原因可能是过高的PGA占比减少了连续相的黏度,因此在乳液测试过程中不足以维持分散相粒子的稳定[17]。
2.3 不同PGA/XG复配比制备S/O/W钙-脂质乳液Zeta-电位
图3为不同PGA/XG复配对制备S/O/W钙-脂质乳液Zeta-电位的影响情况,可以看出不同PGA与XG配比制备的乳液都具有较低的负电荷,这是因为2种阴离子多糖自身带电性质所决定,不同乳液样品电位值无显著差异,这表明所有样品中由电荷产生的颗粒静电斥力相似。

图3 不同PGA/XG复配比制备乳液的Zeta-电位Fig.3 Zeta-potential of emulsions prepared by PGA/XG complex with different ratios
2.4 不同PGA/XG复配比制备S/O/W钙-脂质乳液剪切流变特性
如图4-a所示,不同PGA/XG复配比制备S/O/W钙-脂质乳液的表观黏度均随着剪切速率的增加而减小,显现出剪切稀化现象。这是由于乳液中含有大量絮状颗粒以及XG分子的假塑性,在高剪切速率下乳液中网络结构断裂以及液滴变形所导致[18]。图4-b显示了不同乳液样品的初始表观黏度,随着PGA比例的增加,乳液表观黏度降低,这是因为体系的黏度主要是连续相中XG分子链缠结所形成,过高的PGA比例造成乳液体系黏度值大幅下降,不利于维持乳液的稳定性,这与2.2节的结果一致。
2.5 不同PGA/XG复配比制备S/O/W钙-脂质乳液微观结构
如图5所示,单一XG制备的乳液具有较大的液滴尺寸,PGA与XG复凝聚后制备的乳液液滴均减小,这可能是由于PGA与XG复配后有较好的界面活性,阻碍了油滴间的聚集。复配W相乳液样品随着PGA比例的增加,乳液液滴尺寸先减小后略微增加,这可能是由于更小的液滴具有更大的比表面积,高速分散过程中XG含量不足以覆盖油滴表面所导致[19]。此外,我们可以观察到在部分油滴内部有黑色阴影,推测这是附着在油滴内部的碳酸钙,这与之前报道的有关花青素的研究相类似[20]。

a-复合图像;b-卡尔科弗卢尔荧光增白剂染色W相;c-尼罗红染色O相
2.6 不同PGA/XG复配比制备S/O/W钙-脂质乳液粒径分布
为了更好地佐证CLSM观察到的乳液微观结构,采用S3500激光粒度仪对其进行了粒度分布测量。如图6所示,单一XG制备乳液平均粒径较大,约12.66 μm,且粒径分布曲线峰形较宽。随着PGA的加入,乳液粒径分布曲线变窄,这表明PGA与XG复配制备乳液的液滴大小更加均一。复配W相乳液样品随着PGA比例的增加,乳液粒径分布曲线先向左移后又轻微右移,这表明复配W相乳液样品平均粒径先减小后增加,PGA-XG(4∶6)乳液的平均粒径约1.59 μm,这与激光共聚焦显微镜观察到的结果一致。
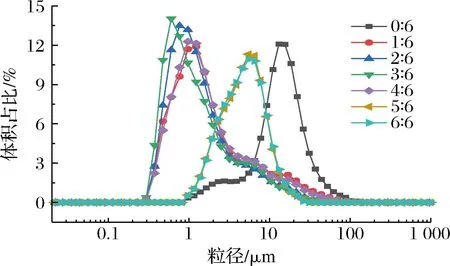
图6 不同PGA/XG复配比制备乳液的粒径分布Fig.6 Particle size distribution of emulsions prepared by PGA/XG complex with different ratios
2.7 PGA-XG复凝聚结构表征
为了进一步探究PGA与XG复合物的结构特性以及相互作用方式,采用激光共聚焦显微镜、扫描电镜、傅里叶变换红外光谱、X射线衍射对复凝聚后的W相进行了表征。
2.7.1 激光共聚焦显微镜
图7显示了不同PGA/XG配比复凝聚后的微观结构。单一的XG分子成链状,PGA与XG复配后,PGA分子基于XG的分子链进行附着,二者发生了相互作用。不同配比下相互作用程度不同,随着PGA含量的增加,二者形成的半互穿网络结构先增强后减弱[21]。这可能是由于不同配比下2种多糖聚合物分子基团平衡结果不同所导致的[22]。

图7 不同PGA/XG复配比CLSM图像Fig.7 CLSM images of PGA/XG complex with different ratios
2.7.2 扫描电镜
基于不同PGA/XG复配比制备乳液的不稳定指数在PGA-XG(4∶6)之后发生徒长,采用扫描电镜对单一PGA、XG和PGA-XG(4∶6)的表观形貌进行了观察。如图8所示,单一XG呈现出有孔洞的云状或片状,且表面相对粗糙;单一PGA呈现出长丝网状。二者复配后呈现出致密的片状结构,孔洞消失且表面光滑。说明二者通过相互作用形成了互补结构。

a-30×;b-100×;c-500×
2.7.3 傅里叶红外光谱


图9 PGA、XG和PGA-XG(4∶6)的傅里叶红外光谱Fig.9 Fourier transform infrared (FTIR) spectra of PGA, XG, and PGA-XG (4∶6) complex
2.7.4 X射线衍射
图10显示了单一XG、PGA、PGA和XG复凝聚后的X射线衍射图谱,可以看出,XG衍射峰为18.67°、30.04°、40.31°,PGA的衍射峰为13.32°、30.04°、40.31 °。2种多糖都显示出较宽的衍射峰,这表明二者都是无定形结构。当PGA与XG复凝聚后,XG的结晶区域减小,结晶峰变窄。这表明二者存在相互作用,PGA的加入打破了XG原有的结构,使得XG结晶度下降。ZHENG等[24]证实原花青素与直链淀粉通过氢键结合,造成了大米淀粉结晶度下降,这与傅里叶红外光谱结果相一致。

图10 PGA、XG和PGA-XG(4∶6)的 XRD图谱Fig.10 X-ray diraction (XRD) patterns of PGA, XG, and PGA-XG (4∶6) complex
3 结论
PGA与XG复配提高了XG在油水界面的表面活性,降低了表面张力,二者以氢键相互作用,进而形成网络结构。以PGA与XG不同复凝聚比为W相,制备了负载碳酸钙的S/O/W钙-脂质乳液,当PGA与XG配比为4∶6时,乳液具有较好的稳定性和流变学特性。微观结构及粒径分布表明,乳液液滴分布均一,碳酸钙位于O相内部。因此,通过S/O/W体系提高了碳酸钙的分散稳定性,实现了食品固相(Solid,S)、油相(Oil,O)与水相(Water,W)的三相均一化。对于纯碳酸钙而言,胃中释放速率较快,可能对胃产生刺激。因此,可在后续研究负载碳酸钙的S/O/W钙-脂质乳液体外模拟消化胃液转化Ca2+动力学以及钙的生物可及性,解析S/O/W钙-脂质乳液的消化特性及控释规律。
猜你喜欢
文萃报·周二版(2023年48期)2023-12-27
汉语世界(2022年5期)2022-10-15
无机盐工业(2021年1期)2021-01-08
石材(2020年12期)2020-12-31
基层中医药(2020年10期)2020-11-27
中成药(2018年5期)2018-06-06
小布老虎(2016年4期)2016-12-01
上海金属(2016年2期)2016-11-23
美食堂(2015年3期)2015-05-30
上海金属(2014年3期)2014-12-19
