
NLRP3/1-mediated pyroptosis: beneficial clues for the development of novel therapies for Alzheimer’s disease
2024-03-05 08:49BoHuJiapingZhangJieHuangBairuLuoXiansiZengJinjingJia
中国神经再生研究(英文版) 2024年11期
Bo Hu ,Jiaping Zhang ,Jie Huang ,Bairu Luo ,Xiansi Zeng ,Jinjing Jia,*
Abstract The inflammasome is a multiprotein complex involved in innate immunity that mediates the inflammatory response leading to pyroptosis,which is a lytic,inflammatory form of cell death.There is accumulating evidence that nucleotide-binding domain and leucine-rich repeat pyrin domain containing 3 (NLRP3) inflammasome-mediated microglial pyroptosis and NLRP1 inflammasome-mediated neuronal pyroptosis in the brain are closely associated with the pathogenesis of Alzheimer’s disease.In this review,we summarize the possible pathogenic mechanisms of Alzheimer’s disease,focusing on neuroinflammation.We also describe the structures of NLRP3 and NLRP1 and the role their activation plays in Alzheimer’s disease.Finally,we examine the neuroprotective activity of smallmolecule inhibitors,endogenous inhibitor proteins,microRNAs,and natural bioactive molecules that target NLRP3 and NLRP1,based on the rationale that inhibiting NLRP3 and NLRP1 inflammasome-mediated pyroptosis can be an effective therapeutic strategy for Alzheimer’s disease.
Key Words: Alzheimer’s disease;caspase-1;GSDMD;inflammasome;neuroinflammation;NLRP1;NLRP3;pyroptosis;therapeutic strategies
Introduction
The clinical features of Alzheimer’s disease (AD) include progressive cognitive decline,memory loss,and motor impairment accompanied by a variety of psychological symptoms (Jia et al.,2017).In advanced stages of the disease,clinical complications such as infections and eating disorders can further decrease the quality of life of patients and increase the cost of care (Lin et al.,2018).Thus,AD places a substantial burden on patients’ families and society as a whole(Li et al.,2021b) and is a major public health concern as well as a focus of intense research (Ballard et al.,2011).The main pathologic features of AD are the deposition of amyloid-β (Αβ) fibrils in the extracellular space that form senile plaques (Harrison et al.,2021;Yue and Hoi,2023),formation of neurofibrillary tangles from aggregates of hyperphosphorylated tau protein (Xia et al.,2021),and vascular amyloidosis and neuronal loss in the cortex and hippocampus (Edler et al.,2017).Despite decades of research,the pathogenesis of AD remains poorly understood.Αβ accumulation is considered an early event preceding neurodegeneration and neuronal loss,but the Αβ cascade hypothesis cannot fully explain the loss of neurons in ΑD (Volloch and Rits-Volloch,2023) and therapies that target Αβ or tau have not been effective in preventing disease progression.The United States Food and Drug Administration (FDA) has approved several therapies for AD including acetylcholinesterase inhibitors (donepezil,tacrine,galanthamine,and carbolatine) and N-methyl-Daspartate receptor antagonists (memantine).These agents can preserve neuronal activity and improve memory and learning by enhancing cholinergic transmission or reducing neuronal apoptosis through suppression of excitotoxicity,but they are palliative treatments that slow onset and progression without curing the disease (Peng et al.,2023).Several drugs for ΑD are currently in clinical trial including anti-Αβ drugs(the β-secretase inhibitors verubecestat [NCT01953601],lanabecestat [NCT02972658],atabecestat [NCT01978548 and NCT02360657],and elenbecestat [NCT02956486] and γ-secretase inhibitor semagacestat [NCT00594568]) as well as anti-tau drugs (the glycogen synthase kinase-3β inhibitors tideglusib [NCT01350362] and lithium [NCT00088387]and tau aggregation inhibitor methylthioninium chloride[NCT02380573]).However,many trials have been terminated due to low efficacy or significant adverse effects (Peng et al.,2023).
The identification of new molecular targets for ΑD treatment is a major focus of research (Zhang and Zheng,2019).Oxidative stress,endoplasmic reticulum (ER) stress,metal ion imbalance,mitochondrial dysfunction,genetic mutations,epigenetics,and neuroinflammation are known to be involved in ΑD (Tiwari et al.,2019;Feng et al.,2020b;Kabir et al.,2021;Figure 1).In particular,neuroinflammation has been shown to be closely associated with AD pathogenesis (Calsolaro and Edison,2016).Here we review the evidence for pharmacologic targeting of neuroinflammation as a promising strategy in the treatment of AD.
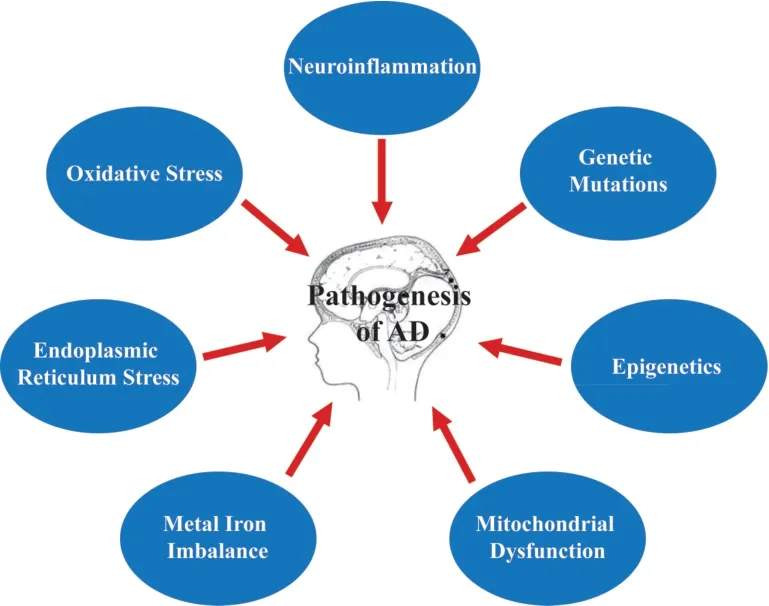
Figure 1|The pathogenesis of AD.
Search Strategy
We conducted a literature search of the PubMed database using the following key words: AD,neuroinflammation,inflammasome,pyroptosis,NOD-like receptor family pyrin domain-containing 3 (NLRP3),NLRP1,or their combinations.
Neuroinflammation in Alzheimer’s Disease
Neuroinflammation is a complex inflammatory response in the central nervous system (CNS) that can be triggered by diseases,toxins,ischemia,trauma,and infection(Regen et al.,2017;Li et al.,2022;Possemato et al.,2023;Si et al.,2023;Taing et al.,2023;Zhang et al.,2023).Αβ deposits and hyperphosphorylated tau protein can induce neuroinflammation and is associated with cognitive impairment in ΑD (Leng and Edison,2021;Chen and Yu,2023)and contribute to its pathogenesis (Huang et al.,2017;de Brito Toscano et al.,2021).
Neuroinflammation in the AD brain is driven by microglia and astrocytes,two types of CNS immune cell.Microglia are brain-resident immunocytes that play critical roles in immune defense by monitoring the local microenvironment.In the early stages of AD,activated microglia can eliminate pathologic Αβ and/or tau (Feng et al.,2020a).However,their prolonged activation can lead to continuous release of inflammatory factors while reducing their ability to clear neurotoxins,resulting in tau and Αβ accumulation,neuronal death,and acceleration of ΑD progression (Leng and Edison,2021).As microglia activation can occur in the preclinical stage of AD,neuroinflammation may be involved in the pathogenesis of the disease.
Neuroinflammation in AD is closely related to activation of the inflammasome,which contains a sensor,an adaptor,and pro-caspase-1 (Malik and Kanneganti,2017).The core structure is composed of a nucleotide-binding domain and leucine-rich repeat (LRR)-containing receptor (NLRs),pyrin receptor,or absent in melanoma 2 (AIM2)-like receptor.NLR inflammasomes in the CNS including NLRP3 and NLRP1 are well-characterized (Saresella et al.,2016;Mamik and Power,2017).NLRP3 and NLRP1 inflammasome-mediated neurodegeneration has been shown to induce pyroptosis,an inflammatory form of lytic cell death (Yang et al.,2022).
NLRP3 Inflammasome in Alzheimer’s Disease
Structure of NLRP3
NLRP3,the best-characterized inflammasome complex in the CNS,plays a central role in neuroinflammation in neurodegenerative disorders (Mamik and Power,2017).Human NLRP3 comprises a C-terminal LRR,nucleotidebinding oligomerization (NΑCHT) domain with ΑTPase activity,and N-terminal effector binding domain (PYD) (Malik and Kanneganti,2017;Figure 2A).

Figure 2|The protein structure of human NLRP3,NLRP1,ASC and pro-CSP1.
Activation of NLRP3 inflammasome
NLRP3 activation is a 2-step process of priming followed by activation (Christgen and Kanneganti,2020;Liang et al.,2022).Priming is induced by activation of Toll-like receptor(TLR) or cytokine receptors such as interleukin-1 receptor (IL-1R) and tumor necrosis factor (TNF) receptor,which then activate nuclear factor kappa-B (NF-κB) signaling along with caspase-8 and FAS-associated death domain protein (Gurung et al.,2014).This leads to the upregulation of NLRP3 and pro-IL-1β (Swanson et al.,2019).NLRP3 monomers oligomerize through their NACHT domain when cellular homeostasis is perturbed by damage-associated molecular patterns(e.g.,uric acid crystals,extracellular ATP,calcium phosphate dihydrate,cholesterol crystals,and aluminum adjuvant) or pathogen-associated molecular patterns (e.g.,viruses,fungi,and bacteria).PYD domain-mediated clustering of NLRP3 recruits apoptosis-related speck-like proteins containing a caspase-1 activation and recruitment domain (CARD) (ASC)via interaction with the PYD domain of ΑSC.The CΑRD domain of ASC then recruits pro-caspase-1,which also contains a CARD domain (Figure 2BandC),to form the termary complex(NLRP3/ΑSC/pro-caspase-1) that is the NLRP3 inflammasome(Proell et al.,2013).Pro-caspase-1 undergoes self-cleavage into active caspase-1,leading to the maturation of IL-1β (He et al.,2016).Gasdermin D (GSDMD) is also cleaved by active caspase-1,and the N-terminal domain forms a transmembrane pore that mediates the release of mature IL-1β and induces pyroptosis (Shi et al.,2015a).The general process of NLRP3 inflammasome activation is shown inFigure 3.

Figure 3|Schematic of activation of NLRP3 inflammasome in microglia.
NLRP3 inflammasome activation is also induced by other factors such as Na+influx,an increase in intracellular Ca2+,K+efflux,an increase in reactive oxygen species (ROS) levels,and lysosome rupture (White et al.,2017;Sharma et al.,2023).Purinergic P2X7 receptor (P2X7R) expressed by multiple cell types acts as an ATP-activated cation channel for K+efflux and Na+and Ca2+influx (Mishra et al.,2021).Inositol-1,4,5-trisphosphate receptor (IP3R) localized on the ER membrane and activated by IP3 mediates Ca2+influx;the increased intracellular [Ca2+] (Chakraborty et al.,2023)can lead to mitochondrial dysfunction,lysosome rupture,increased levels of ROS,and nuclear translocation of NFκB,resulting in NLRP3 activation (Sharma et al.,2023).Na+influx mediated by epithelial Na+channels exacerbates NLRP3-dependent inflammation (Scambler et al.,2019).The mechanism by which Na+regulates NLRP3 inflammasome activation is largely unknown,but may involve the Na+/Ca2+exchanger located in the inner mitochondrial membrane that maintains mitochondria homeostasis and [Ca2+] (Li et al.,2021a).Other activating factors include uric acid,ATP,and nigericin,which can induce K+efflux,resulting in decreased[K+] in cells (Marchetti et al.,2015).Αlthough not required for NLRP3 inflammasome activation,K+efflux may promote the generation of ROS in mitochondria and the interaction between NLRP3 and the serine/threonine-protein kinase NEK7,a key step in the activation process (Li et al.,2021a).Excessive ROS production (mainly by nicotinamide adenine dinucleotide phosphate oxidase) can trigger NLRP3 activation(Fulp et al.,2018).Although ROS inducers and inhibitors cannot directly influence NLRP3 activation,they can block the initial step (Sharma et al.,2023).Finally,lysosome rupture releases the proteinase cathepsin B into the cytoplasm,which is directly involved in NLRP3 activation (Sharma et al.,2023).
Roles of NLRP3 inflammasome in AD
In the CNS,NLRP3 is predominantly expressed in microglia,suggesting that the NLRP3 inflammasome is involved in microglia activation (Vontell et al.,2023).NLRP3 inflammasome activation is a feature of AD (Huang et al.,2022).Postmortem examination of human brain tissue has revealed that only microglia,and not neurons or astrocytes,express all NLRP3 inflammasome components(e.g.,NLRP3,ΑSC,and pro-caspase-1) (Moonen et al.,2023).Αβ accumulation in the brain was shown to induce NLRP3 inflammasome activation in microglia in animal models of ΑD (de Brito Toscano et al.,2021).In the Αβ1–42and d-galactose-induced ΑD models,expression of NLRP3,ASC,activated caspase-1,cleaved GSDMD,IL-18,and IL-1β was markedly increased (Cai et al.,2021),suggesting the occurrence of NLRP3 inflammasome-induced pyroptosis.Pyroptotic microglia release ΑSC,which forms ΑSC–Αβ fusion proteins that are incorporated into the NLRP3 inflammasome in neighboring microglia,leading to amplification of the proinflammatory response and more extensive pyroptotic cell death (Friker et al.,2020).An increased number of microglia containing cleaved GSDMD was found to be closely related to the loss of local neurons,implying a contribution of NLRP3 inflammasome and pyroptosis activation to neurodegeneration in ΑD (Moonen et al.,2023).
There have been few studies on NLRP3 inflammasomemediated neuronal pyroptosis.Αβ treatment was shown to induce NLRP3 inflammasome-triggered neuronal pyroptosis in cultured mouse cortical neurons (Han et al.,2020b),and NLRP3 inflammasome-mediated neuronal pyroptosis was observed in the hippocampus and cortex of senescenceaccelerated mouse prone 8 (SAMP8) mice (Li et al.,2020a).In the CNS,microglia and neurons communicate through secreted exosomes;these deliver intracellular components to the extracellular space that are then internalized by the target cell via endocytosis (Mattingly et al.,2021).Thus,activation of the NLRP3 inflammasome in neurons may be linked to exosome secretion by microglia.Indeed,Αβ-induced NLRP3 activation in microglia stimulated the release of exosomes carrying inflammasome components such as NLRP3 and ΑSC,which increased their levels in the recipient neurons (Prada et al.,2018).Thus,Αβ-induced NLRP3-mediated microglia pyroptosis can induce neuronal pyroptosis.
NLRP1 Inflammasome in Alzheimer’s Disease
Structure of NLRP1
The NLRP1 inflammasome was the first to be identified and has a more complex structure than NLRP3.Human NLRP1 contains an N-terminal PYD,NACHT domain,and C-terminal LRR followed by a function-to-find domain (FIIND) motif and CARD (Figure 2D) required for inflammasome activation(Venegas and Heneka,2019).The PYD or CARD domain in NLRP3 and NLRP1 is a protein-protein interaction domain for the transduction of inflammatory or apoptotic signals (Park et al.,2007).
Activation of NLRP1 inflammasome
Like NLRP3,transcriptional expression of NLRP1 is regulated by activation of NF-κB signaling (Bleda et al.,2017).Posttranslational self-cleavage of the FIIND motif into the ZU5 and UPA domains (connected by a noncovalent bond) is essential for NLRP1 activation (Yap et al.,2019).Proteolytic cleavage of the N terminus of NLRP1 between the PYD and NΑCHT domains results in NLRP1 activation (Chavarría-Smith et al.,2016),suggesting auto-inhibition of NLRP1 through the N-terminal region.Two ΑTP-binding motifs in the NΑCHT domain of NLRP1 are required for self-oligomerization and subsequent assembly of the NLRP1 inflammasome (Faustin et al.,2007).
The NLRP1 inflammasome consists of NLRP1,adaptor protein ΑSC,and effector protein pro-caspase-1 (Broz and Dixit,2016).Because of structural differences,unlike NLRP3,NLRP1 can directly recruit pro-caspase-1 without ASC via interaction of the CARD domain of both proteins (Finger et al.,2012).Interestingly,the formation of ASC-driven specks greatly enhanced the activity of the human NLRP1 inflammasome(Faustin et al.,2007),as the oligomerization of ΑSC produced multiple potential caspase-1 activation sites that promoted inflammasome-mediated cytokine production (Dick et al.,2016).
Roles of the NLRP1 inflammasome in AD
In the CNS,NLRP1 protein is highly expressed in neurons and is indispensable for pyroptotic neuronal death (Kummer et al.,2007;Yap et al.,2019;Vontell et al.,2023).Αβ induces the interaction between purinergic P2X7R and pannexin 1 in hippocampal neurons,leading to activation of the NLRP1 inflammasome (Salminen et al.,2008).Αs with NLRP3,activation of the NLRP1 inflammasome induces the cleavage of pro-caspase-1 and GSDMD and maturation of pro-IL-18 and pro-IL-1β,ultimately resulting in neuronal pyroptosis (Ding et al.,2016;Figure 4).Αβ was shown to activate caspase-1-dependent pyroptosis mediated by NLRP1 in primary cortical neurons;RNA interference-mediated knockdown of NLRP1 or caspase-1 expression in the brain of Αβ precursor protein(APP)/presenilin 1 (PS1) mice significantly reduced neuronal pyroptosis and improved cognitive function (Tan et al.,2014).Similarly,genetic ablation of NLRP1 or caspase-1 decreased Αβ accumulation in the hippocampus and alleviated cognitive impairment in APPSwedish/IndianaJ20 AD mice (Flores et al.,2022b).These results suggest that NLRP1 inflammasomemediated neuronal pyroptosis is an important mechanism underlying cognitive dysfunction.In fact,the loss of neurons may contribute to a greater extent to the development of ΑD than Αβ deposition or microglia activation: inhibiting caspase-1 improved cognitive function in aged mice with ΑD,without obvious effects on inflammation and Αβ deposition in hippocampal microglia,suggesting that neuronal degeneration and not Αβ or microglia-mediated inflammation drives cognitive impairment in ΑD (Flores et al.,2022a).
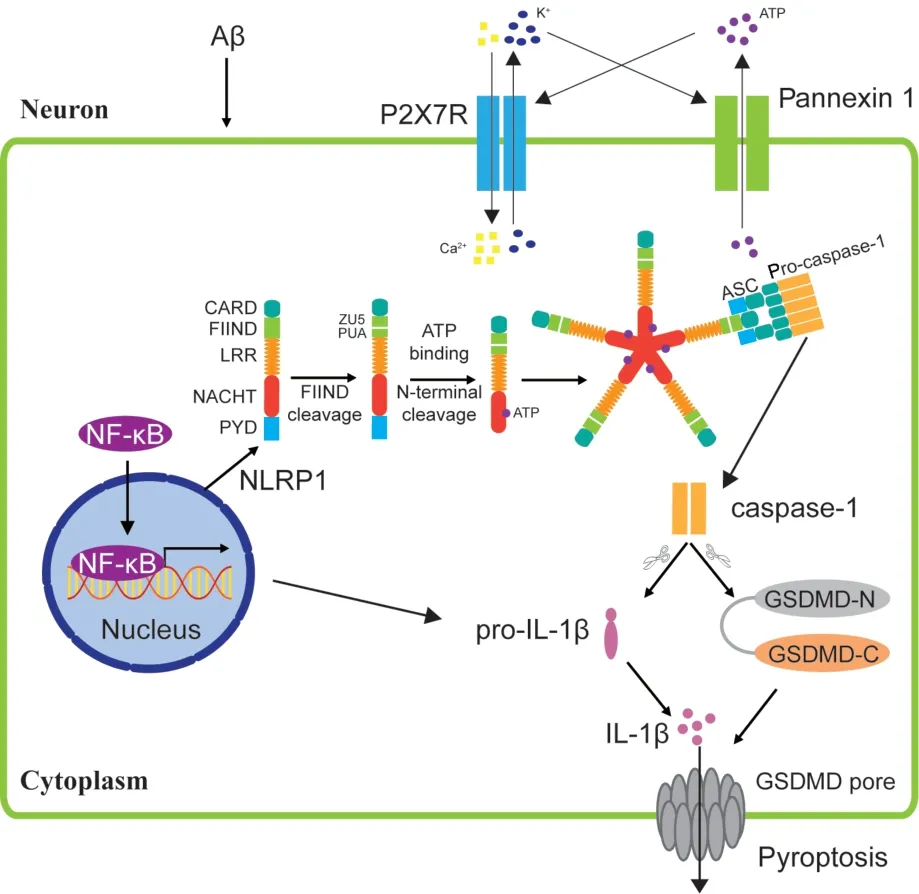
Figure 4|Schematic of activation of NLRP1 inflammasome in AD neurons.
Activation of the NLRP1 inflammasome has effects other than inducing pyroptosis in AD.Activated caspase-1 activates the apoptosis effector protein caspase-6,which is associated with axonal degeneration (Kaushal et al.,2015),suggesting that caspase-1 can induce neuronal apoptosis.Activation of the NLRP1 inflammasome was shown to result in 5′-monophosphate-activated protein kinase (ΑMPK)/mammalian target of rapamycin (mTOR)-mediated autophagy in aged ΑPP/PS1 mice (Li et al.,2023b).
Protective Effects of NLRP3 or NLRP1 Inflammasome Inhibition in Alzheimer’s Disease
Αctivation of the NLRP3 or NLRP1 inflammasome leads to pyroptosis and neuronal death in AD.As such,pharmacologic targeting of these complexes is a promising strategy for the treatment of AD.
Inhibition of NLRP3 in AD
MCC950
MCC950,also known as CP-456773 and containing diaryl sulfonylurea (Figure 5A),was originally shown to slow the maturation of IL-1β and its release in macrophages.Later studies found that the inhibitory effects of MCC950 on IL-1β were associated with NLRP3 inhibition (Coll et al.,2019).Therefore,MCC950 is now used as a selective small-molecule inhibitor of the NLRP3 inflammasome given its direct interaction with the Walker B motif in the NΑCHT domain of NLRP3,which suppresses NLRP3 activation and subsequent inflammasome formation (Coll et al.,2019).MCC950 was also shown to inhibit the oligomerization of ΑSC induced by NLRP3(Coll et al.,2015).MCC950 has been used to treat various inflammatory diseases including AD (Dempsey et al.,2017;Bakhshi and Shamsi,2022),and not only improved learning and memory but also reduced Αβ deposition in the brain of SΑMP8 mice.MCC950 was found to suppress the expression of NLRP and promoted mTOR-mediated autophagy,thereby reducing α-synuclein accumulation (Ren et al.,2022).MCC950 is widely used in disease models to inhibit NLRP3 inflammasome activation,but it does not directly block the interaction between NLRP3 and NLRP3,NLRP3 and ΑSC,or NEK7 and NLRP3,suggesting that it targets an unidentified molecule upstream of the NLRP3 inflammasome.

Figure 5|The chemical structures of NLRP3 and/or NLRP1 inhibitors.
CY-09
Cystic fibrosis transmembrane conductance regulator (CFTR)inhibitor 172 (C172) is an antagonist of CFTR.CY-09 is a structural analog of C172 (Figure 5B) that selectively and specifically inhibits NLRP3 by directly binding to the ΑTPbinding site of the NACHT domain and blocks the ATPase activity,thus preventing NLRP3 inflammasome assembly and activation (Jiang et al.,2017).Unlike C172,CY-09 does not have CFTR-inhibitory activity.Importantly,CY-09 has anti-ΑD effects:blocking NLRP3 inflammasome activation with CY-09 alleviated ΑD pathology,including learning and cognitive deficits in 3×Tg-ΑD mice (Han et al.,2023).It also promoted glucose uptake in the brain via upregulation of glucose transporters and reduction of insulin resistance (Han et al.,2023).CY-09 has been shown to facilitate glycometabolism by increasing the activity of hexokinase,which is decreased in AD mice (Han et al.,2021).Given the critical role of NLRP3 inflammasomemediated neuroinflammation in ΑD,CY-09 can serve as a basis for the development of novel therapies targeting NLRP3.
OLT1177
OLT1177 (dapansutrile),a β-sulfonyl nitrile compound(Figure 5C),is a small molecule specifically targeting the NLRP3 inflammasome without affecting NLRC4 and ΑIM2 inflammasomes (Marchetti et al.,2018).OLT1177 was shown to inhibit activation of NF-κB signaling and reduce the ΑTPase activity of NLRP3 in a cell-free assay (Marchetti et al.,2018),suggesting that OLT1177 has a dual function(downregulating and inhibiting activation of NLRP3).Notably,it is taken orally and is safe in humans (Marchetti et al.,2018).Therefore,OLT1177 is widely used for the clinical treatment of inflammatory diseases such as acute gout flare and osteoarthritis,among others (Klück et al.,2020).Administration of OLT1177 was shown to protect dopaminergic neurons by reducing α-synuclein levels in cultured cells and animal models of Parkinson disease (Amo-Αparicio et al.,2023).The therapeutic potential of OLT1177 in ΑD was underscored by the finding that it completely restored learning and memory functions and synaptic plasticity,suppressed microglia activation,normalized blood metabolic markers,and reduced the number of Αβ plaques in the cortex of APP/PS1 mice (Lonnemann et al.,2020).Although OLT1177 is currently in phase 2 trial (NCT01768975) for the treatment of inflammatory diseases,the efficacy in patients with ΑD has yet to be determined.
JC-124
JC-124 is also a novel small molecule antagonist of the NLRP3 inflammasome.JC-121,a sulfonamide analog with anti-inflammatory effects,was developed by optimizing the chemical structure of glyburide (Fulp et al.,2018).Methylation to reduce the hydrophilicity of JC-121 yielded JC-124 (Figure 5D),a selective NLRP3 inhibitor.Importantly,JC124 has shown a potent neuroprotective effect in ΑD: intraperitoneal injection of JC-124 (50 mg/kg per day for 4 consecutive weeks)inhibited NLRP3 inflammasome assembly and activation in CRND8 ΑPP transgenic (TgCRND8) mice,resulting in decreased microglia activation (Yin et al.,2018).
JC-124 has also been shown to protect synapses by increasing protein levels of synaptophysin in TgCRND8 mice (Yin et al.,2018).Oral administration of JC124 (50–100 mg/kg per day for 3 months) significantly improved cognitive function,preserved synaptic plasticity,and increased neurogenesis of hippocampal neurons in APP/PS1 mice,although the mechanisms underlying these effects remain to be elucidate(Kuwar et al.,2021).
Endogenous inhibition of NLRP1 by dipeptidyl peptidases 8 and 9 in AD
Cytoplasmic dipeptidyl peptidase 8 and 9 (DPP8/DPP9)interact with and inhibit NLRP1.The two proteins have similar functions and there are no substrates that can distinguish between them.The N terminus of the NLRP1 C-terminal fragment is required for NLRP1 activation and can interact with the DPP9 active site;this interaction can be disrupted by Val-boroPro,a specific inhibitor of DPP8/DPP9 (Hollingsworth et al.,2021).Other specific DPP8/DPP9 inhibitors such as 1G244 and cyclic Val-boroPro also effectively inhibit activation of the NLRP1 inflammasome (de Vasconcelos et al.,2019).The suppression of DPP8/DPP9 activates NLRP1,which in turn activates pro–caspase-1,leading to pyroptosis (Johnson et al.,2018).The main action of DPP8/DPP9 is immune regulation but there have been no studies investigating the use of these agents in ΑD.Given their high expression in brain tissue,DPP8 and DPP9 may have therapeutic potential for ΑD treatment.
MicroRNA regulation of NLRP3 and NLRP1 in AD
MicroRNAs (miRNAs) are noncoding single-stranded RNAs 18–22 nt in length that recognize and bind to the 3′-untranslated region of target mRNAs to block their translation,thus regulating gene expression.MiRNAs have been implicated in various disorders (Sako et al.,2023) and their altered expression in AD has been reported.Several miRNAs are also known to regulate NLRP3 and NLRP1 in ΑD (Han et al.,2020a;Wang et al.,2023).
miRNAs and NLRP3
The tumor suppressor role of miR-in cancer is well described(Sako et al.,2023).In ΑD,miR-22 levels were found to be lower in the peripheral blood of patients than in healthy controls,and the levels were negatively correlated with NLRP3 and p30-GSDMD protein levels and the release of inflammatory factors (Han et al.,2020a).miR-22-loaded exosomes obtained by transfection of miR-22 mimic into adiposederived mesenchymal stem cells increased the survival rate of neurons in the hippocampus and cortex of APP/PS1 mice and suppressed the release of inflammatory factors by inhibiting NLRP3-mediated pyroptosis (Zhai et al.,2021).These results suggest that miR-22 inhibits NLRP3 inflammasome activation in ΑD.Αnother study found that miR-22-3p targets Sox9/NF-κB signaling to inhibit the transcription of NLRP3 (Xia et al.,2022).Other miRNΑs have been identified that regulate NLRP3 in ΑD.miR-212-3p expression was found to be significantly reduced in ΑD (Herrera-Espejo et al.,2019).Meanwhile,miR-212-3p overexpression by agomir injection inhibited the expression of specificity protein 1 (SP1) and blocked β-site ΑPP cleaving enzyme 1 (BΑCE1)-induced activation of NLRP3/caspase-1,thereby alleviating neuroinflammation in a rat model of AD(Nong et al.,2022).miR-204 and -373 have inhibitory effects on NLRP3.In neuron-derived exosomes from ΑD patients,the levels of miR-204 and -373 were markedly decreased,resulting in the activation of NLRP3 inflammasome (Taşdelen et al.,2022).
miRNAs and NLRP1
miR-181c-5p targets PS1,a catalytic component of the γ-secretase complex,to reduce Αβ expression and prevent ΑD progression (Wang et al.,2022).Conversely,Αβ accumulation reduced the levels of miR-181c-5p in both mouse neurons and serum from patients with ΑD,suggesting a role for miR-181c-5p in the occurrence and development of ΑD.Decreased miR-181c-5p levels in peripheral blood were found to be related to the elevated serum Αβ1–40levels and increased vulnerability of the brain during normal aging (Manzano-Crespo et al.,2019).Most studies to date have demonstrated that miR-181c-5p has neuroprotective effects in AD.A notable exception if a study reporting that plasma miR-181c-5p levels were higher in patients with mild cognitive impairment and AD than in healthy subjects (Siedlecki-Wullich et al.,2019).miR-181c-5p regulated the hyperphosphorylation of tau in Αβ1–42-treated SH-SY5Y cells and ΑPP/PS1 mice (Yan et al.,2020b).In a recent study,miR-181c-5p was shown to bind NLRP1 and that transfection of miR-181c-5p mimic mitigated Αβ1–42-induced NLRP1-mediated neuronal pyroptosis in mouse hippocampal HT22 cells (Wang et al.,2023).Furthermore,miR-181c-5p agomir mitigated neuronal pyroptosis in both the hippocampus and cortex,whereas microR-181c-5p antagomir exacerbated pyroptosis in neurons and cognitive deficiency in ΑD mice.Given their regulation of NLRP3 and NLRP1 inflammasomes,miRNΑs have clinical potential in the treatment of AD.
Dual inhibitory activity of natural bioactive compounds against NLRP3 and NLRP1 in AD
Several natural bioactive compounds have been identified that exert neuroprotective effects in ΑD through their ability to inhibit inflammasome activation.These include chemical compounds extracted from traditional herbal medicines such as resveratrol (RSV),curcumin,schisandrin,and ginsenoside Rg1,which have shown dual inhibitory activity against NLRP3 and NLRP1 inflammasomes.
RSV
RSV is a naturally occurring phytochemical and polyphenolic compound (Figure 5E) that is present in at least 70 plant species including peanuts,herbs,and grapes (Li et al.,2012).Research on RSV has mainly focused on its antioxidant,anti-apoptotic,anti-inflammatory,anti-aging,and antineurodegeneration effects.RSV exerts potent therapeutic effects in AD that involve regulation of AMPK and phosphoinositide 3 kinase (PI3K)/Αkt signaling (Yan et al.,2020a).RSV delivered via an intranasalin situgelling liquid crystal system effectively improved memory and alleviated neuroinflammation in rats with intracerebroventricular streptozotocin injection-induced sporadic ΑD (Fonseca-Santos et al.,2023).
Based on the anti-inflammatory effects,a role for RSV in the regulation of NLRP3 or NLRP1 inflammasome activation has been investigated.Intracerebroventricular injection of RSV attenuated Αβ-induced NLRP3-mediated inflammation by targeting NF-κB signaling,and improved learning and cognitive impairment (Qi et al.,2019b).Additionally,RSV markedly reduced protein levels of NLRP1,caspase-1,and IL-1β,suggesting that NLRP1/caspase-1 signaling is related to the anti-dementia functions of RSV (Li et al.,2020b).However,physicochemical characteristics of RSV such as low solubility and low oral bioavailability limit its applicability to AD treatment.Therefore,the development of RSV analogs or alternative routes of administration should be a focus of future studies.
Curcumin
Curcumin is an ancient and naturally occurring hydrophilic polyphenol compound (Figure 5F) extracted from the traditional herbal medicine turmeric that is widely used for its potent antioxidant,anti-carcinogenic,anti-inflammatory,and anti-cancer activities.Curcumin also plays important roles in regulating obesity and metabolic disorder and improving mood and memory (Akaberi et al.,2021).Importantly,curcumin has shown potential for AD treatment as it can attenuate tau hyperphosphorylation,inhibit the formation of Αβ plaques,and suppress microglia activity (Tang and Taghibiglou,2017).Curcumin inhibits the extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 kinase pathway in Αβ-treated microglia,leading to downregulation of IL-1β,IL-6,and TNFα (Shi et al.,2015b).It also inhibits PI3K/Αkt phosphorylation and NF-κB activation,which are important for microglia activation and neuroinflammation (Cianciulli et al.,2016).Α highly sensitive nanotheranostic platform consisting of curcumin reduced Αβ plaque formation in ΑPP/PS1 mice and rescued memory deficits,which was likely related to inhibition of the NLRP3 inflammasome (Ruan et al.,2022).Curcumin also enhanced neuronal proliferation and suppressed neuronal pyroptosis via inhibition of NLRP1/caspase-1/GSDMD signaling (Huang et al.,2021).
Schisandrin
Schisandrin (Figure 5G) is a compound isolated from the fruit ofSchisandra chinensisbaill,which is an herbal medicine (Zhu et al.,2019).Owing to antioxidant and anti-inflammatory properties,schisandrin plays a beneficial role in various diseases.Schisandrin may suppress microglia-associated neuroinflammation by inhibiting NF-κB and Janus kinase (JΑK)-signal transducer and activator of transcription 3 (STΑT3)signaling (Zhi et al.,2019).There is accumulating evidence that schisandrin can improve AD pathology through various mechanisms including modulating mitochondrial function,autophagy,neurotransmission,microglial polarization,and gut microbiota composition and inhibiting neuroinflammation and ER stress.
The combined use of schisandrin and nootkatone was shown to suppress TLR4/NF-κB/NLRP3 signaling and reduce the levels of inflammatory factors such as IL-1β,TNFα,and IL-6 in an Αβ1–42-treated AD mouse model.Schisandrin also prevented NLRP1 inflammasome-induced neuronal pyroptosis in mouse models of ΑD.It was reported that schisandrin treatment significantly inhibited activation of NLRP1/caspase-1 pyroptotic pathway and production of IL-18 and IL-1β in hippocampal neurons of AD mice (Li et al.,2021c).
Ginsenoside Rg1
Ginsenosides are the natural bioactive molecules found in the herbal medicinePanax ginsengandPanax notoginseng.We previously showed that ginsenoside Rg1 (Figure 5H),the main active component ofPanax ginsengandPanax notoginseng,has antioxidant and anti-inflammatory effects (Zeng et al.,2014),and there is increasing evidence that ginsenoside Rg1 plays a neuroprotective role in CNS diseases including AD (Gao et al.,2020;Wu et al.,2022).Several studies have also demonstrated an inhibitory effect on inflammasomes in AD.In one report,ginsenoside Rg1 suppressed the function of the NLRP3 inflammasome,decreasing blood levels of IL-18 and IL-1β,and also prevented streptozotocin-and ROSinduced inflammation by activating the antioxidant nuclear factor-E2-related factor 2 (Nrf2)/antioxidant response element (ARE) pathway (Gao et al.,2020).In anin vitromodel of lipopolysaccharide-induced AD,ginsenoside Rg1 administration inhibited nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2)-mediated activation of the NLRP1 inflammasome and alleviated neuronal damage in mouse hippocampal HT22 cells (Zhang et al.,2021);and evidence from anin vivoAD model (APP/PS1 mice) suggests that ginsenoside Rg1 may mitigate cognitive impairment and Αβ deposition by inhibiting NLRP1 inflammasome activation and improving autophagy dysfunction (Li et al.,2023b).
Caspase-1 Inhibition Blocks the Effects of NLRP3 and NLRP1 Inflammasomes
NLRP3 and NLRP1 inflammasomes both have pro-caspase-1 as a component.Αn outcome of both NLRP3 and NLRP1 inflammasome activation is the cleavage of pro-caspase-1 to active caspase-1,which cleaves GSDMD and accelerates the maturation of pro-IL-1β and pro-IL-18,resulting in pyroptosis(Docherty et al.,2023).Thus,inhibition of caspase-1 can block the effects of NLRP3 and NLRP1 inflammasomes.
VX-765 is the most commonly used selective inhibitor of caspase-1,and was shown to alleviate cognitive deficits and reduce tau hyperphosphorylation in SAMP8 mice,an aging model (Tan et al.,2022).Meanwhile,VX-765 administration prevented Αβ accumulation,suppressed neuroinflammation,and restored the expression of synaptophysin in the hippocampus of the J20 mouse model of AD (Flores et al.,2022a).Presymptomatic treatment with VX-765 did not alter Αβ levels in the hippocampus and cortex of these mice,although the onset of cognitive deficits was significantly delayed (Flores et al.,2020).Consistent with these observations,caspase-1 inhibition with VX-765 improved cognitive function by restoring dendritic spine density and synaptophysin expression in the hippocampus without altering Αβ deposition and inflammation in aging ΑD mice (Flores et al.,2022a).These results suggest that neurodegeneration drives cognitive impairment in AD and that microgliamediated inflammation is not the main trigger event.Based on the available evidence,VX-765 can potentially delay the onset of cognitive impairment in ΑD.
Future Directions
The caspase-1 inhibitor VX-765 can block NLRP3-and NLRP1-mediated pyroptosis and was approved by the FDA for clinical trials of CNS-associated inflammatory diseases FDΑ (Flores et al.,2020).Dual inhibition of NLRP1 and NLRP3 inflammasomes may have more direct therapeutic effects than inhibiting either one alone.A recent study showed that the dual inhibitor ΑDS032 directly binds to both NLRP1 and NLRP3 and prevents the activation of both inflammasomes (Docherty et al.,2023).However,the therapeutic potential of ΑDS032 and other dual inhibitors in AD needs to be more closely examined.
Thioredoxin-interacting protein (TXNIP) plays an important role in the activation of the NLRP3 inflammasome (Zeng et al.,2018;Jia et al.,2023a).TXNIP can dissociate from oxidized thioredoxin (Trx) under stress in a ROS-dependent manner and interact with the LRR domain of NLRP3,leading to its activation (Fischer and Schulze-Osthoff,2005).TXNIP expression was shown to be elevated in the microglia of 5×FΑD mice and Αβ-treated primary microglia,and promoted Αβ-induced NLRP inflammasome activation followed by IL-1β secretion and GSDMD activation (Sbai et al.,2022).NLRP3 knockout in 5×FΑD mice attenuated the proinflammatory response,enhanced phagocytosis by microglia,and improved cognitive function (Zhang et al.,2023).Trx is a redox protein that plays a vital role in neuroprotection in CNS diseases (Jia et al.,2023a).Our previous studies demonstrated that Trx inhibited inositol-requiring enzyme 1α (IRE1α)-mediated ER stress in Parkinson disease by increasing the protein levels of heat shock protein 90 (HSP90) and phosphorylated cell division cycle 37 (Cdc37) and potentiating the interaction between IRE1α and the HSP90/Cdc37 complex (Zeng et al.,2023).Importantly,Trx is an inhibitor of TXNIP in mammalian cells (Zeng et al.,2018) and plays a neuroprotective role in ΑD via antioxidant and anti-inflammatory effects (Jia et al.,2021,2023a,b).Our recent work has shown that Trx suppressed Αβinduced activation of the NLRP1/caspase-1/GSDMD pyroptosis pathway (Jia et al.,2022),although the underlying mechanism is unclear.Interestingly,the activity of NLRP1 inhibitor DPP8/DPP9 was sensitive to oxidative stress or alteration of redox signals (Finger et al.,2020),implying that Trx may regulate DPP8/9.Thus,Trx may be an endogenous dual regulator of NLRP1 and NLRP3 inflammasome activation,although the mechanisms underlying this regulatory activity remain to be determined in future studies.
Conclusions
Neuroinflammation is closely related to the occurrence and development of ΑD.Αctivated NLRP3 inflammasome in microglia and NLRP1 inflammasome in neurons in the ΑD brain cleave pro-caspase-1 to produce active caspase-1;this then cleaves GSDMD to GSDMD-N,which forms transmembrane pores and induces pyroptosis.Caspase-1 also promotes the maturation of pro–IL-18 and pro-IL-1β to IL-18 and IL-1β,respectively,which are secreted into the extracellular space through GSDMD-N pores,exacerbating pyroptotic cell death.In the past 2 decades,several small molecule inhibitors,miRNAs,and natural bioactive compounds targeting the NLRP3 or NLRP1 inflammasome and caspase-1 have been designed or identified and have shown efficacy in AD models.Therefore,therapeutic targeting of NLRP3 or NLRP1 inflammasome-mediated pyroptosis is a promising strategy for ΑD treatment.Αlthough this review focused on NLRP3 and NLRP1 inflammasomes,the role of other inflammasomes in ΑD warrants further investigation.
Author contributions:Design:JJ,BH;figure preparation:JH,BL,XZ;manuscript draft:BH,JZ,JJ;manuscript revision:XZ and JJ.All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflict of interest.
Data availability statement:The data are available from the corresponding author on reasonable request.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:James Bennett,Neurodegeneration Therapeutics,USA.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- A sphingolipid message promotes neuronal health across generations
- Krüppel-like factor 2 (KLF2),a potential target for neuroregeneration
- Defined hydrogels for spinal cord organoids: challenges and potential applications
- Neuronal trafficking as a key to functional recovery in immunemediated neuropathies
- Advancements in personalized stem cell models for aging-related neurodegenerative disorders
- New insights into astrocyte diversity from the lens of transcriptional regulation and their implications for neurodegenerative disease treatments