
糖足散质量控制方法的初步研究
2024-03-04 08:32:14徐璐娜周玉芳阮洪生王翰华王又迪
人参研究 2024年1期
徐璐娜,周玉芳,阮洪生,陈 云,王翰华,王又迪
(1.浙江药科职业大学中药学院,宁波 315503;2.浙江医院药剂科,杭州 310013)
糖尿病足是老年糖尿病患者致残的严重慢性并发症之一[1]。现阶段中医对糖尿病足的体外治疗,主要采取的处理方法有中药足疗熏洗、中药外敷涂抹等[2-4]。糖足散是浙江医院中医科应用多年的协定处方,由红花等9 味中药组成。方中红花、当归、牛膝温通散寒、活血化瘀,合为君药。桂枝、醋乳香、细辛辛温通络,配合君药散寒活血,三药为臣。赤芍,清热凉血,发挥佐制之用。凤仙透骨草、桑枝引诸药透入经络、血脉,通达四肢而祛风、活血、止痛。糖足散熏洗患者足部能扩张血管内径,加快患肢血液运行,改善足部微循环,用糖足散协助弥可保治疗早期糖尿病足,临床疗效显著[5]。本实验对糖足散进行了初步的定性定量研究,为其质量标准的完善与提升提供依据。
1 仪器与材料
1.1 仪器
BK6000 型显微镜(重庆奥特光学仪器有限公司);Good Look-2000 型薄层色谱成像系统(上海科哲生化科技有限公司);UV-6100 型紫外可见分光光度计(上海元析仪器有限公司);FA2004 型电子分析天平(上海恒平科学仪器有限公司);HH-2 型恒温数显水浴锅(常州国华电器有限公司);HY-08 型高速粉碎机(北京环亚天元机械技术有限公司)。
1.2 材料
当归、红花、牛膝对照药材(批号:120927-201617、120907-201412、121066-201809),β-蜕皮甾酮、人参皂苷Ro 对照品(批号:111638-201706、111903-201604)均购自中国食品药品检定研究院;芦丁对照品(批号:B20771,纯度98%)购自上海源叶生物科技有限公司;中药饮片均购自杭州华东饮片有限公司;葡萄糖标准溶液1000 mg/L、3,5-二硝基水杨酸试液(Phygene);娃哈哈纯净水;其余试剂均为分析纯。
2 方法与结果
2.1 糖足散样品制作
称取当归、红花等中药饮片适量,分别粉碎成粉末(过3 号筛),按糖足散处方中比例(当归、赤芍、牛膝、桂枝、透骨草各18 g、桑枝24 g、红花12 g、醋乳香10 g、细辛6 g)混匀,即得糖足散样品。
2.2 显微鉴别[6]
取糖足散样品粉末适量,制片、观察,并描述其显微特征。本品粉末红棕色,花粉粒类圆形或椭圆形,直径约60μm,外壁具有短刺和点状雕纹,3 个萌发孔(红花);石细胞呈类圆形、类方形或类长方形,壁一面菲薄(桂枝),或壁厚,胞腔小(桑枝);草酸钙方晶,直径5~20μm(桑枝),草酸钙簇晶,直径15~30μm(赤芍);不规则团块无色或淡黄色,表面及周围扩散出众多细小颗粒,久置溶化(醋乳香);非腺毛由多个细胞构成,部分细胞内含棕色物质(凤仙透骨草)。详见图1。
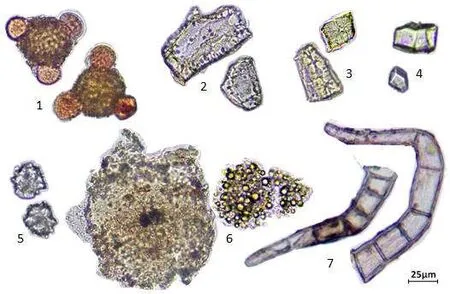
图1 糖足散显微特征(40×)
2.3 薄层鉴别
2.3.1 阴性样品制作
按“2.1”项下方法,分别制作缺当归、缺红花、缺牛膝、缺赤芍、缺桂枝、缺细辛的阴性样品。
2.3.2 当归[6]
取糖足散样品4 g(含当归0.5 g),加乙醚50 mL,回流提取1 h,取滤液,低温挥干,加乙醇1 mL 溶解残渣,即得供试品溶液。取缺当归的阴性样品3.5 g,当归对照药材0.5 g,按上述方法分别制备阴性样品溶液、对照药材溶液。吸取上述溶液各5 μL,按序点于同一硅胶G 薄层板,展开系统选正己烷-乙酸乙酯(4∶1),于365 nm 紫外光灯下观察。
2.3.3 红花[6]
取糖足散样品5.9 g(含红花0.5 g),加80%丙酮溶液20 mL,超声20 min,取续滤液,即得供试品溶液。取缺红花的阴性样品5.4 g,红花对照药材0.5g,按上述方法分别制备阴性样品溶液、对照药材溶液。吸取上述溶液各20 μL,按序点于同一硅胶H 薄层板,展开系统选乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4),于日光下观察。
2.3.4 牛膝[6]
取糖足散样品10 g(含牛膝1.3 g),加80%甲醇100 mL,回流提取4 h,取滤液,蒸干,加温水5 mL 溶解残渣,转移至D101 型大孔吸附树脂柱,用水、20%乙醇各100 mL 先后洗脱柱子(弃洗脱液),再用80%乙醇100 mL 洗脱,收集、蒸干洗脱液,加80%甲醇1 mL 溶解残渣,即得供试品溶液。取缺牛膝的阴性样品8.7 g、牛膝对照药材1.3 g,按上述方法分别制备阴性样品溶液,对照药材溶液。再取β-蜕皮甾酮对照品、人参皂苷Ro 对照品,加甲醇分别制成0.5 mg/mL 的对照品溶液。吸取上述溶液各15 μL(对照品溶液10 μL),点于同一硅胶G 薄层板,展开系统选二氯甲烷-甲醇-水-甲酸(14∶6∶1∶0.1),5%香草醛硫酸溶液喷淋,105℃加热,斑点呈色清晰后观察。
2.3.5 薄层鉴别结果
当归、红花、牛膝薄层色谱图中,主斑点清晰,阴性无干扰。详见图2~4。
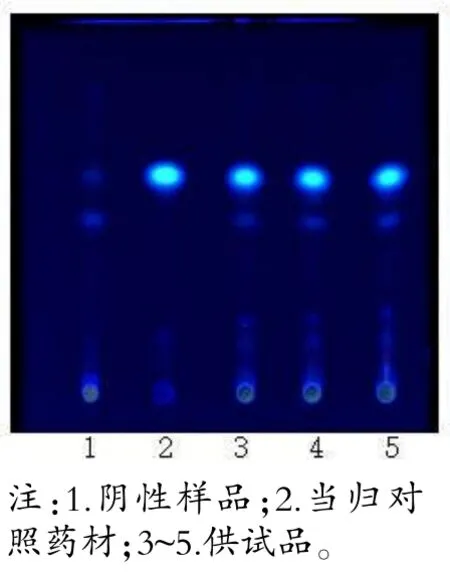
图2 当归薄层色谱
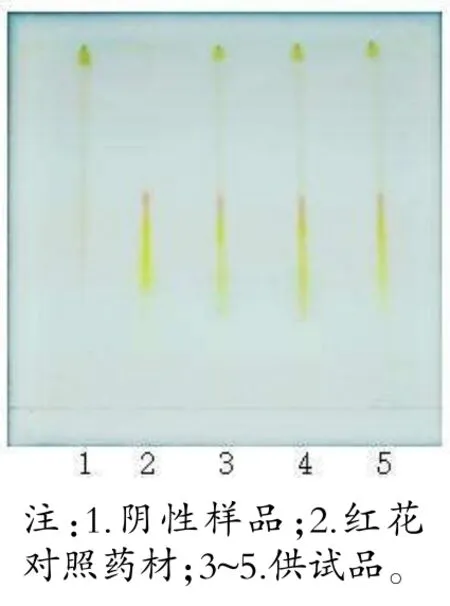
图3 红花薄层色谱
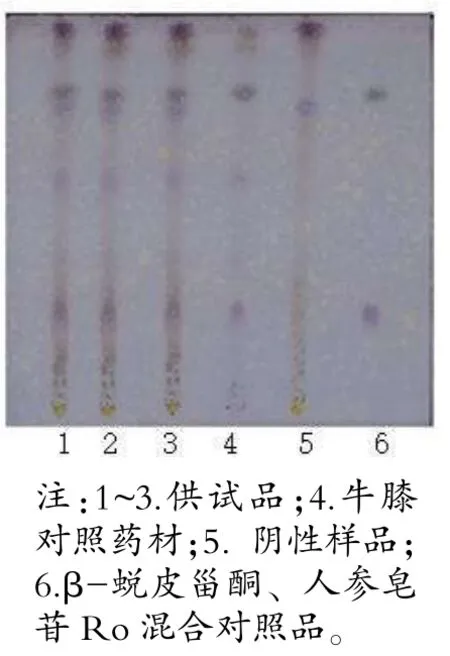
图4 牛膝薄层色谱
2.4 糖足散总黄酮含量测定
2.4.1 对照品溶液制备
取芦丁对照品(纯度:98%)11.5 mg,精密称定,置50 mL 容量瓶中,加70%乙醇40 mL,置水浴上微热使溶解,放冷,加70%乙醇至刻度,摇匀,即得(质量浓度为0.2254 mg/mL)。
2.4.2 供试品溶液制备
取糖足散3 g,精密称定,置250 mL 锥形瓶中,加水135 mL(料液比1∶45),浸泡30 min,水浴加热回流提取0.5 h,提取液减压抽滤,滤液转移至250 mL 容量瓶中,加水定容,摇匀,即得。
2.4.3 样品测定
精密移取待测溶液2mL 于25mL 容量瓶中,加水至6mL,加5%亚硝酸钠溶液1mL,混匀,放置6 min,加10%硝酸铝溶液1mL,混匀,放置6min,加氢氧化钠试液10mL,再加水至刻度,摇匀,放置15min。以相应试剂为空白,按照紫外-可见分光光度法[6],在310nm波长下检测。
2.4.4 检测波长的选择
精密量取“2.4.1~2.4.2”项下对照品溶液3 mL、供试品溶液2 mL,分别置于25 mL 容量瓶中,按“2.4.3”项下方法,自“加水至6 mL”开始,依法操作,在250~600 nm 波长范围内进行扫描,发现在310 nm 波长处有较大的吸收且信号较稳定。综合考虑后,最终选用310 nm 作为本实验最终检测波长。
2.4.5 正交试验考察水提工艺
在水浴工作状态下,设计四因素三水平L9(34)正交试验,考察料液比(1∶25、1∶35、1∶45)、浸泡时间(30、60、90 min)、提取温度(80、90、100℃)、提取时间(0.5、1.0、1.5 h)对糖足散中总黄酮含量的影响,最终以直观分析结果结合节能高效的原则,确定了最佳提取参数,即液料比1∶45,浸泡时间30 min,提取温度100℃,提取时间0.5 h。
2.4.6 线性关系考察
精密移取“2.4.1”项下对照品溶液1、2、3、4、5、6 mL 分别置于25 mL 容量瓶中,制成质量浓度为9.02、18.03、27.05、36.06、45.08、54.10 μg/mL 的标准溶液,分别按“2.4.3”项下方法测定,用吸光度、对照品质量浓度分别作纵、横坐标,得回归方程:Y=0.019 4X+0.007 5(R2=0.999)。芦丁在9.02~54.10 μg/mL 范围内线性关系良好。
2.4.7 精密度试验
精密移取芦丁对照品溶液3 mL,按“2.4.3”项下方法,重复测定6 次。结果对照品溶液吸光度RSD 为0.39%,表明该仪器精密度良好。
2.4.8 重复性试验
精密移取同一份供试品溶液6 份,每份2 mL,按“2.4.3”项下方法测定吸光度,计算得到总黄酮百分含量RSD 为1.82%,表明该方法重复性良好。
2.4.9 稳定性试验
分别精密移取对照品溶液3 mL、供试品溶液2 mL,按“2.4.3”项下方法操作,分别于0、10、20、30、40、50 min 测定。结果对照品溶液、供试品溶液吸光度RSD 分别为1.82%、0.80%,表明50 min 内上述两种溶液稳定性较好。
2.4.10 加样回收率试验
称取已知含量的糖足散粉末0.15g,共6 份,置于25 mL 锥形瓶中,分别加入芦丁对照品适量,加水13.5 mL,浸泡30 min,水浴加热回流提取0.5 h,提取液减压抽滤,滤液转移至25 mL 容量瓶中,加水定容,摇匀,即得供试品溶液,测定吸光度,计算回收率。结果见表1。
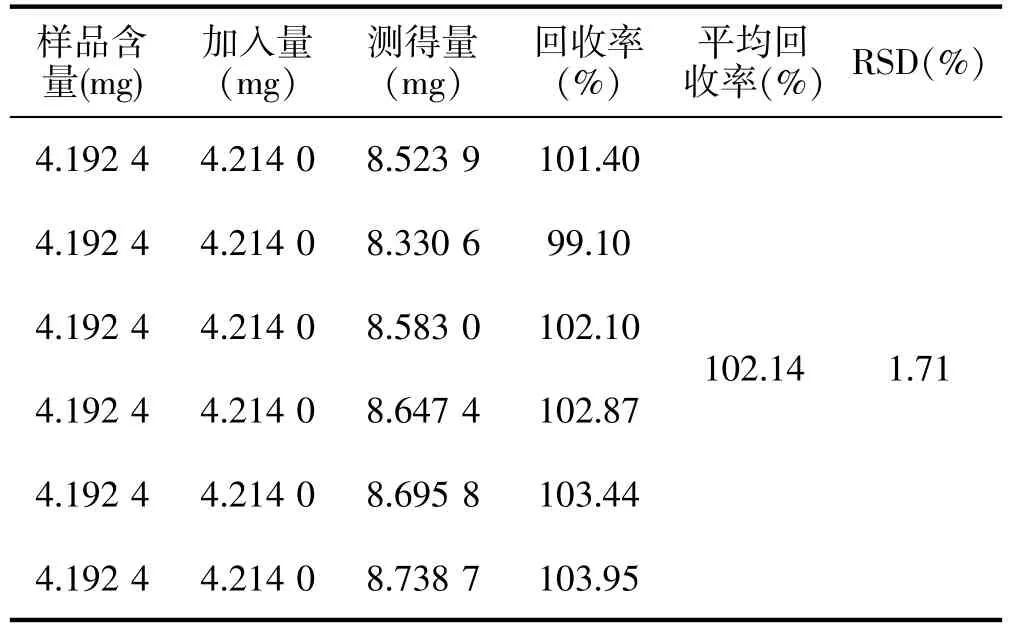
表1 总黄酮回收率结果(n=6)
2.4.11 样品含量测定
称取3 g 糖足散粉末3 份,按“2.4.2”项下供试品溶液制备法制备,按“2.4.3”项下方法操作测定。结果样品中总黄酮平均含量为3.042%(RSD=2.00%)。
2.5 糖足散多糖含量测定
2.5.1 对照品溶液的制备
取葡萄糖标准溶液(1000 mg/L),按需求稀释。
2.5.2 供试品溶液的制备
2.5.2.1 单糖供试品溶液的制备
称取糖足散50 mg 于50 mL 容量瓶中,加超纯水适量,振摇使溶解,用超纯水定容至刻度,摇匀,用直径0.80 μm 的滤膜滤过,取续滤液即得,备用。
2.5.2.2 总糖供试品溶液的制备
称取糖足散50 mg 于100 mL 磨口三角瓶中,先加盐酸(1→2)20 mL,置沸水浴中10 min,取出,静置,待放冷后用40%氢氧化钠溶液调节pH 值至中性,然后移入100 mL 的容量瓶中,用纯净水稀释,定容至刻度,摇匀,用直径为0.80 μm 滤膜滤过,取续滤液25 mL 至50 mL 容量瓶中,用纯净水稀释,定容至刻度,摇匀即得,备用。
2.5.3 样品测定
精密量取待测溶液4mL,置于20mL 具塞比色管中,加入DNS 试液2mL,摇匀,放置沸水浴中加热7min,取出后放入冰水中冷却至室温,加纯净水6mL,摇匀,平行作空白,按照紫外分光光度法[6],在515nm波长下检测。
2.5.4 检测波长的选择
分别精密量取“2.5.1~2.5.2”项下对照品溶液适量、单糖溶液与总糖溶液各4 mL,置于20 mL 的具塞刻度试管中,分别按照“2.5.3”项下方法,自“加入DNS试液2mL”开始,依法操作,在470~700 nm 波长范围内进行光谱扫描。结果显示三种溶液在515 nm 左右波长下有最大吸收。综合考虑后,选用515 nm 作为检测波长。
2.5.5 DNS 用量的考察
取5 支20 mL 具塞比色管,分别精密加入葡萄糖对照品溶液1 mL,补纯净水至4 mL,分别准确加入DNS 试液1、2、3、4、5 mL,放入沸水浴中加热5 min,取出后立即用冰水浴冷却至室温,加纯净水稀释,定容至刻度,摇匀后测定各自吸光度。详见图5。当用量为2 mL 时,吸光度已达最大值。
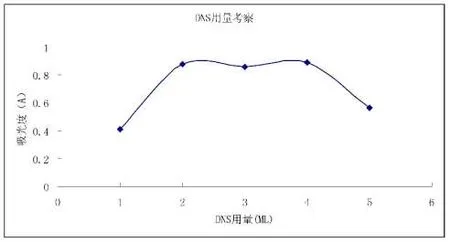
图5 DNS 用量与吸光度的关系
2.5.6 反应时间考察
精密量取葡萄糖对照品溶液1 mL 至20 mL 比色管中,分别补纯净水至4 mL,加入DNS 试液2 mL,分别放入沸水浴中1、3、5、7、9 min,取出后立即用冰水浴冷却至室温,加纯净水稀释,定容至刻度,摇匀后测定各自吸光度。详见图6。由图6 可知,显色反应时间为7 min 时,测定的吸光度值达到最大,故选7 min为本实验最佳显色反应时间。
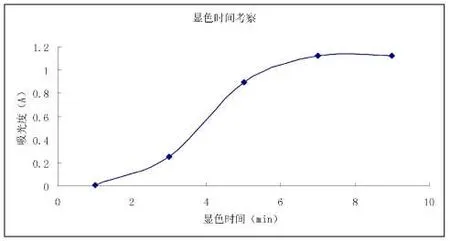
图6 显色时间与吸光度的关系
2.5.7 标准曲线绘制
分别精密吸取葡萄糖标准溶液(1000 mg/L)1、1.5、2、2.5、5、6.25mL 于25mL 容量瓶中,依次加纯净水稀释,定容至刻度,摇匀,即得。精密量取不同浓度对照品溶液各4 mL,按照“2.5.3”项下方法测定,以葡萄糖浓度为横坐标,吸光度A 为纵坐标,回归方程为:Y=5.112 9X-0.200 5(R2=0.999 8),表明葡萄糖在0.04~0.25 mg/mL 范围内线性关系良好。
2.5.8 精密度试验
精密量取葡萄糖标准溶液(1000 mg/L)2.85 mL,置于25 mL 容量瓶中,加纯净水稀释,定容至刻度,得所需对照品溶液。按样品测定方法重复测定6 次。结果对照品溶液吸光度RSD 为0.18%,表明该仪器精密度良好。
2.5.9 稳定性试验
分别精密量取对照品溶液(按精密度试验项下的对照品溶液配制)、单糖溶液、总糖溶液各4 mL,按照样品测定方法测定,于0~50 min 内每隔10 min 测定1 次,结果各组溶液吸光度RSD 值均≤2%,可认为3种溶液在50 min 内稳定。
2.5.10 重复性试验
精密移取同一份单糖供试品溶液6 份,每份4 mL,分别置于20 mL 的具塞比色管中,按照样品测定方法测定,总糖供试品溶液的测定操作同上。结果单糖溶液吸光度RSD 为1.92%;总糖溶液吸光度RSD为1.58%,显示该方法重复性良好。
2.5.11 加样回收率试验
2.5.11.1 单糖加样回收率试验
对照品溶液配制:精密量取葡萄糖标准溶液(1000 mg/L)1.70 mL,置于25 mL 容量瓶中,加纯净水稀释,定容至刻度,摇匀即得,备用。配制糖足散单糖溶液6 份,分别移取2 mL 单糖样品溶液置于20 mL 的具塞比色管中,分别加入2 mL 对照品溶液,按照样品测定方法测定。测得单糖平均回收率为98.19%,RSD 为2.32%,结果见表2。
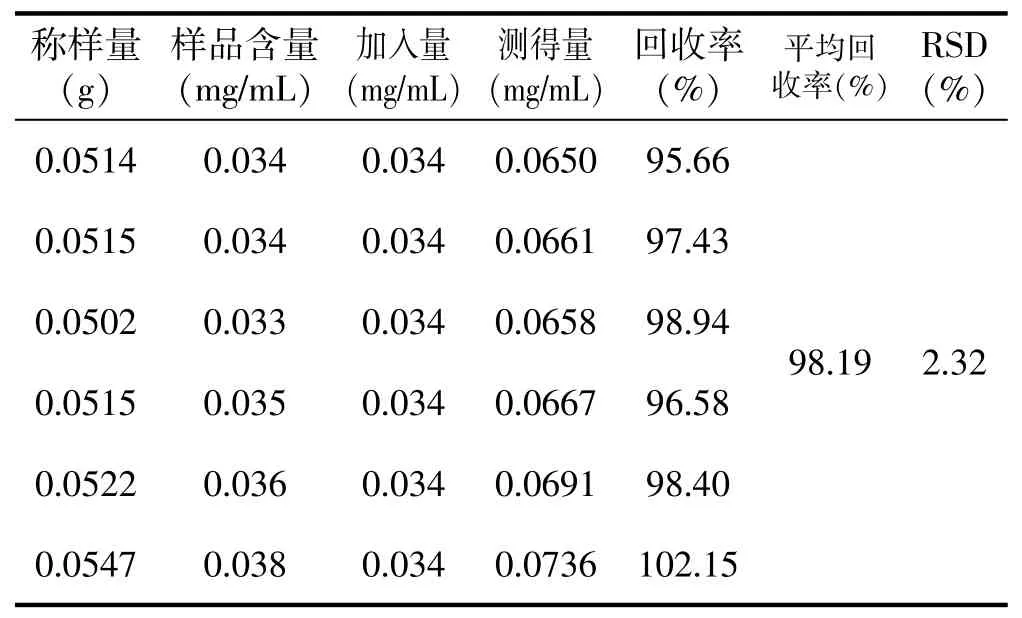
表2 单糖回收率结果(n=6)
2.5.11.2 总糖加样回收率试验
对照品溶液配制:精密量取葡萄糖标准溶液(1000 mg/L)2.85 mL,置于25 mL 容量瓶中,加纯净水稀释,定容至刻度,摇匀即得,备用。配制糖足散总糖溶液6 份,分别移取2 mL 总糖样品溶液置于20 mL 的具塞比色管中,分别加入2 mL 对照品溶液,按照样品测定方法测定。测得总糖平均回收率为103.03%,RSD 为1.77%,结果见表3。

表3 总糖回收率结果(n=6)
2.5.12 样品多糖含量测定
按“2.5.2”项下分别制备单糖供试品溶液和总糖供试品溶液各6 份,按照样品测定方法测定,依法显色,测定吸光度并计算各自含量,再按“多糖含量=(总糖含量-单糖含量)×0.9”计算多糖含量。结果样品中单糖的平均含量为7.470%(RSD=3.00%),总糖的平均含量为46.423%(RSD=2.79%),代入上式,算得多糖含量为35.058%。
3 讨论
显微鉴别中,透化技术的灵活运用是镜下观察清晰与否的关键。试验中发现对于只需微透化的样品,滴水合氯醛试液后,可尝试略过加热步骤,放置片刻,待其自然透化后再行观察,有时得到的效果会更佳。另外,因糖足散中当归的纺锤形韧皮薄壁细胞相对不易找到,故暂未纳入本文。
薄层鉴别中发现环境温湿度对薄层展开有一定影响。试验前期,牛膝薄层展开环境温度过高,分离不理想,后改在阴凉室中进行,分离度便有所改善。湿度会影响硅胶薄层板的吸附能力,建议使用前对硅胶板进行活化,使板内水分含量保持相对一致,尽量减少对Rf 值重现性的影响。此外牛膝薄层中还发现,因供试品中含较多水溶性杂质,不可贪图方便,省略过柱洗脱环节,否则易出现斑点粘连,各组分难以分离的情况。
黄酮类化合物是一类重要的天然产物,糖足散中多味药材富含黄酮类成分,具抗炎、抑菌等作用[7-9],对治疗糖足溃疡有一定的疗效,故将总黄酮作为评价糖足散质量的重要指标之一。测定黄酮类化合物含量的方法常见的有高效液相色谱法、紫外分光光度法,但高效液相色谱法更适合单体化合物的分离,因此,采用紫外分光光度法测定糖足散中总黄酮的含量[10]。
糖足散中多味药材含多糖类成分,有抗炎抑菌、抗氧化、抗凝血等活性,可能是治疗糖尿病足的有效组分,故把多糖作为糖足散的一个质控指标[11-14]。目前测定多糖的比色法有很多,常见的是蒽酮-硫酸比色法和苯酚-硫酸比色法,但这两种方法存在缺陷,因单糖类杂质也会与苯酚或蒽酮反应,致使测定结果偏高[15]。另外蒽酮-硫酸比色法,其稳定性受测定条件影响较大(如蒽酮试剂不易保存)[16]。DNS 法测定结果受杂质干扰较小,可排除单糖等杂质带来的误差,且DNS 试剂稳定性较好,因而选择DNS 法测定糖足散多糖[17]。
本文通过定性定量分析,对糖足散质量控制方法进行了初步研究,为其质量标准的制定奠定基础,也为临床安全用药提供保障。
猜你喜欢
军事文摘(2023年18期)2023-10-31 08:10:30
世界科学技术-中医药现代化(2022年9期)2023-01-17 07:36:18
军事文摘(2022年10期)2022-06-15 02:30:00
军事文摘·科学少年(2022年5期)2022-06-11 07:18:48
童话世界(2019年14期)2019-06-25 10:11:52
天然产物研究与开发(2019年1期)2019-03-01 05:41:20
中成药(2018年6期)2018-07-11 03:01:16
中学生阅读(初中版)(2016年11期)2017-01-13 01:06:40
天然产物研究与开发(2016年1期)2016-06-05 10:29:25
中南民族大学学报(自然科学版)(2015年2期)2015-12-16 12:11:10
