
PI3K/AKT/mTOR信号通路在肝癌中的作用机制研究进展
2024-03-01 11:40戴依哲李娅楠尚润泽
肝胆胰外科杂志 2024年2期
戴依哲,李娅楠,尚润泽
1.海军军医大学第三附属医院 肝外四科,上海 200433;2.华侨大学附属海峡医院 普通外科,福建 泉州 362000;3.南京大学医学院附属金陵医院 麻醉科,江苏 南京 210023
原发性肝癌包括肝细胞癌(hepatocellular carcinoma,HCC)和肝内胆管细胞癌(intrahepatic cholangiocarcinoma,iCCA),是世界范围内第3大肿瘤致死病因[1]。由于多数肝癌患者首次确诊时已是中晚期,无法行根治性手术切除,因此预后常较差。当前针对进展期肝癌的药物治疗主要包括分子靶向治疗、免疫治疗以及靶向联合免疫治疗等。近年来,随着对肝癌发生发展机制研究的不断深入,人们发现了包括TP53、CTNNB1、ARID1A/2、AXIN1、TSC1/2、PIK3CA在内的肝癌常见的突变基因以及MET、MAPK、Wnt/β-catenin和PI3K/AKT/mTOR等异常激活的关键信号通路[2],这些发现为肝癌的靶向治疗提供了更多可能。PI3K/AKT/mTOR信号通路在细胞的增殖、分化、生存和代谢等多项生命活动中发挥重要的调控作用,也是包括肝癌在内的多种肿瘤中最常被激活的信号通路之一[3]。本文就PI3K/AKT/mTOR信号通路在肝癌发生、发展中的作用机制以及相关分子靶向药物的最新研究进展做一综述。
1 PI3K/AKT/mTOR信号通路的构成
1.1 PI3K/AKT的构成
磷脂酰肌醇3-激酶(phosphoinositide 3-kinase,PI3K),可分为Ⅰ型、Ⅱ型和Ⅲ型三种亚型,其中对Ⅰ型的研究最为广泛。Ⅰ型PI3K由p110和p85组成,分为ⅠA、ⅠB两种亚型。目前研究主要集中在由p85α/β/γ调节亚基及其p110α/β/δ/γ催化亚基组成的具有双重活性的ⅠA型PI3K[4]。PI3K可通过两种途径激活:一是通过胞外多种配体与细胞表面PTK受体结合而激活,二是通过Ras蛋白与催化亚基结合而激活(图1)。
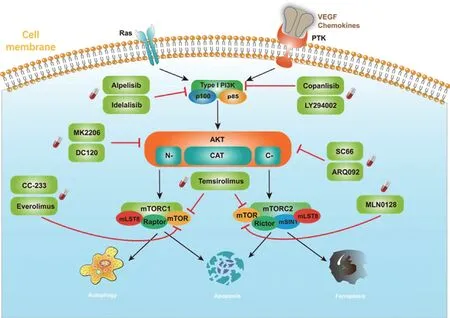
图1 肝癌状态下PI3K/AKT/mTOR通路的构成及相关靶向药物作用位点
蛋白激酶B(protein kinase B,AKT),可分为AKT1、AKT2和AKT3三种亚型,均由3个结构域组成。N端是PH结构域;中间是中央激酶结构域CAT,该结构域具有AKT活化所必需的三磷酸腺苷结合位点;C端是具有调节疏水基序列的延伸域,内有另一个必需位点,在PI3K的激活下可转变为磷酸化的AKT再进一步激活后续位点(图1)。
1.2 mTOR的构成
人类的哺乳动物雷帕霉素靶蛋白复合物(mammalian target of rapamycin,mTOR)由1号染色体短臂(1p36.2)编码,分子量289 kDa,是PI3K蛋白激酶家族的成员[5]。mTOR广泛参与了细胞生长和代谢的多种信号通路[6],它能够整合来自三大营养物质和细胞外信号的输入;mTOR激活后会引起多个下游靶点的磷酸化。mTOR信号级联的激活需要形成三元复合体mTORC1(mTOR复合体-1)和mTORC2(mTOR复合体-2),二者发挥不同的功能[7-8](图1)。
mTORC1是由mTOR、Raptor和mLST8组成的复合体。其中mTOR是复合体的催化亚基,Raptor是mTOR调节相关蛋白,起募集mTORC1底物的作用,还负责识别部分mTORC1底物。三元复合体通过S6激酶/核糖体蛋白S6(RPS6)和真核翻译起始因子-4E结合蛋白-1(eIF4EBP1)级联调节蛋白质合成、葡萄糖和脂质代谢以及细胞自噬,并在肿瘤发生中发挥关键作用[9]。
mTORC2 则是由mTOR、mLST8、Rictor和mSIN1 组成的复合体。同样,mTOR在复合体中起催化亚基作用,而Rictor是对雷帕霉素不敏感的mTOR伴侣蛋白,可以调节AKT、血清/糖皮质激素调节激酶(SGK)和蛋白激酶C等AGC家族蛋白成员。mSIN1通过其N端嵌入到Rictor中,然后围绕mLST8折叠。mSIN1中间保守区域对于mTORC2的底物招募十分重要,而mSIN1的C端PH结构域是mTORC2在膜上定位所必需的(图1)。
2 PI3K/AKT/mTOR通路在肝癌发生发展中的作用
2.1 促进细胞增殖,抑制细胞凋亡
PI3K/AKT/mTOR通路可能通过多种机制促进肿瘤的发生发展。首先,通路中的PI3K可激活AKT为磷酸化AKT,曾文涛等[10]研究显示:磷酸化AKT在HCC中高表达并可进一步磷酸化激活Caspase-9、Bad、FOXO等蛋白促进细胞增殖,抑制细胞凋亡。此外,磷酸化AKT还可以解除TSC1/2 和Rheb对mTOR的抑制作用,使mTOR与Raptor结合后组成mTORC1复合体,进一步磷酸化激活核糖体S6激酶,进而干预5’-TOP结构的mRNA翻译,促进肿瘤细胞的增殖。同时磷酸化mTOR还可以与Rictor结合并通过形成mTORC2复合体磷酸化激活AKT,进一步促进细胞增殖。
c-Myc是多种肿瘤发生发展的重要调控基因,PI3K/AKT/mTOR通路在c-Myc诱导的肝癌中也发挥了重要作用。Xu等[11]在小鼠体内肝癌模型中发现mTORC2在c-Myc驱动的小鼠HCC中被激活,进而导致AKT1被磷酸化激活,从而促进肝癌发展,通过使用抑制剂同时抑制mTORC1/2的表达可以显著抑制小鼠肝癌的生长。
2.2 抑制铁死亡,促进肝癌发生
肝脏是人体中储存铁元素的器官,具有调节体内铁含量的功能,然而过多的铁在肝脏中沉积会导致一系列的问题。2012 年Dixon等[12]首次报道了一种新型的铁离子依赖性的氧化死亡方式——铁死亡。Yi等[13]的研究揭示了PI3K的激活突变或PTEN的失活能够引起包括肝癌细胞在内的多种肿瘤细胞产生铁死亡抵抗,而通过抑制PI3K/AKT/mTOR信号通路能够诱导肿瘤细胞铁死亡,进一步的机制研究发现:通路中mTORC1的持续激活能够通过上调固醇调节元件结合蛋白1(serpine mRNA binding protein 1,SERBP1)的表达引起癌细胞对铁死亡产生抵抗。另一项研究发现PI3K/AKT/mTOR信号通路介导了MCF2L对肝癌铁死亡的负调控作用[14]。
2.3 与乙肝病毒相互作用,促进肝癌发生
肝炎病毒感染是肝癌发生的重要危险因素之一,在感染HBV的患者之中,HBV与PI3K/AKT/mTOR信号通路复杂交互,共同参与了HBV相关肝癌的发生。Zhu等[15]的研究首次发现乙肝病毒HBx蛋白诱导的AFP表达能够通过激活PI3K/mTOR信号传导促进肝细胞的恶性转化。此外,乙肝病毒Pre-S蛋白突变体可以通过内质网应激来激活VEGF-A/VEGFR-2/AKT/mTOR通路,进而促进肝细胞增殖以及后续的恶性转化[16]。另外,Chen等[17]发现乙肝病毒感染相关的PDzk1蛋白可通过调节PI3K/AKT通路和脂肪酸代谢发挥致癌作用。
2.4 参与肝细胞癌的代谢重编程
在癌细胞中,糖酵解产生的大部分丙酮酸离开线粒体并在乳酸脱氢酶(LDH/LDHA)的催化下产生乳酸。Zhang等[18]的研究揭示mTORC1可以抑制NEAT1_2 的表达和paraspeckles小体的形成,并释放RNA结合蛋白NONO和SFPQ,进而与剪接体内的U5 结合,激活mRNA剪接和关键糖酵解酶的表达,这一系列改变导致葡萄糖转运增强和乳酸的产生,并促进体内、外肝癌细胞的生长。非编码RNA与PI3K/AKT/mTOR通路的相互作用也影响着糖代谢重编程,Zhang等[19]研究发现miR-873可以通过靶向NDFIP1蛋白调节AKT/mTOR通路,从而引发糖代谢重编程。此外,PI3K/AKT/mTOR通路还能以多种机制促进肿瘤的脂质合成。SREBP-1是调节细胞脂质稳态的关键基因,它能够调控多种参与脂肪酸合成、甘油三酯合成以及胆固醇合成相关的关键基因的表达[20],而SREBP-1的成熟与激活主要受到PI3K/AKT/mTORC1的调控[21]。另外,有研究发现AKT还可以通过抑制GS3K和Insig2的表达间接上调SREBP-1的表达水平,从而促进肿瘤脂肪酸合成[22-23]。除了参与肝癌的糖、脂代谢重编程外,PI3K/AKT/mTOR同样参与了支链氨基酸的代谢。Ericksen等[24]通过转录组、酶和代谢组学分析发现:在体内、外抑制支链氨基酸分解代谢酶(BCKDHA、ACADS和ACADSB)的活性会引起肝癌组织中支链氨基酸的蓄积,同时增强mTORC1活性和细胞增殖速率。除支链氨基酸外,在谷氨酰胺代谢过程中也发现了PI3K/AKT/mTOR通路的激活,Li等[25]报道了其在肝癌细胞中通过下调GOT2 的表达能够重新编程谷氨酰胺代谢来支持细胞抗氧化系统,从而促进谷氨酰胺分解、核苷酸合成和谷胱甘肽合成,在此过程中PI3K/AKT/mTOR通路被激活,进而促进肝癌的进展。
2.5 介导肝癌血管生成
肿瘤血管新生在肝癌的进展中发挥着重要的作用,近年来的研究发现PI3K/AKT/mTOR通路可以通过多种机制介导肿瘤新生血管的形成。例如,Li等[26]发现,肝癌细胞外泌体来源的长链非编码RNA SNHG16可以通过激活PI3K/AKT/mTOR通路促进肝癌血管生成。Chen等[27]研究表明,在非酒精性脂肪肝相关的肝癌患者中,由于存在脂代谢紊乱以及低氧环境,引起HIF-2α的表达上调,并通过激活PI3K/AKT/mTOR通路促进肿瘤血管生成,而在阻断该通路后肿瘤血管生成明显减少。此外,有研究表明,低氧环境下MAPK通路与PI3K/AKT通路的共同激活也是导致肿瘤血管生成的原因之一[28]。除了经典的肿瘤血管新生外,血管拟态也是另外一种维持肿瘤高代谢特征的生物学行为,PI3K/AKT通路受到CD276(B7-H3)的调控,通过调节下游多种基质金属蛋白酶(MMPs)的表达,参与血管拟态的发生[29]。
2.6 在肝癌耐药中的作用
目前的肝癌治疗指南建议将奥沙利铂作为基础的全身静脉化疗用于治疗不适宜手术切除或局部治疗的晚期和转移性肝癌[30]。但是在治疗过程中部分患者会出现奥沙利铂耐药。研究发现,奥沙利铂处理后的不同肝癌细胞系中PI3K/AKT/mTOR通路均存在不同程度被激活,而PI3K抑制剂Gedatolisib(PKI-587)可以通过抑制DNA损伤修复途径以及抑制PI3K/AKT/mTOR通路活性使耐药的肝癌细胞对奥沙利铂重新致敏[31]。通过抑制PI3K/AKT通路可以减少因线粒体-溶酶体串扰造成的肝癌顺铂耐药[32]。有研究表明,肝癌细胞中的岩藻糖基转移酶家族(fucosyltransferases,FUTs)可以通过调控PI3K/AKT通路上调多重耐药蛋白(multidrug resistance protein 1,MRP1)的表达,造成肝癌细胞对多重药物的耐药[33]。此外,PI3K/AKT/mTOR通路的激活在肝癌靶向药物的耐药中也发挥了重要的作用。研究表明,PI3K/AKT/mTOR通路的激活是肝癌产生索拉非尼耐药的一个关键机制,使用PI3K、mTOR双重抑制剂BEZ235可以通过抑制PI3K/AKT/mTOR通路介导的自噬提高肝细胞癌对索拉非尼的敏感性[34]。
2.7 参与肝癌的肿瘤微环境调控
肝癌的肿瘤微环境除了肿瘤细胞本身,还包括结构成分(如细胞外基质等)、基质细胞(如免疫细胞、成纤维细胞、内皮细胞等)和信号成分(趋化因子、细胞因子、激素和生长因子等),它们共同在肝癌的发生发展中发挥重要作用[35]。在正常肝组织中,细胞外基质主要是由肝星状细胞(hepatic stellate cell,HSC)分化为成纤维细胞后分泌[36]。在肝细胞癌中HSC也作为肿瘤微环境成分之一调控肿瘤的增殖和恶性进展,Liu等[37]研究发现刺激胞外的基质硬度可以促进肝细胞癌的生长以及转移,其机制是通过转录因子E2F3促进HSC分化为具有促瘤作用的成纤维细胞,并且可以通过FGFR1 调控下游PI3K/AKT及MEK/ERK通路。基质细胞成分中的肿瘤相关巨噬细胞(tumor associated macrophage,TAM)在肿瘤发生、转移、血管生成和适应性免疫抑制中发挥关键作用,TAM可分为M1型和M2型,前者具有促炎和抗肿瘤的特点,而后者的功能与前者相反[38-39]。TREM蛋白是一个在髓系细胞中表达的免疫球蛋白细胞表面受体家族蛋白,在巨噬细胞中下调TREM1蛋白的表达可以通过抑制PI3K/AKT/mTOR通路促进M2型TAM向M1型转化,从而改变肝癌微环境的组成,抑制肿瘤进展[40]。CXCL5是趋化因子CXC亚家族成员之一,Zhou等[41]发现CXCL5可以通过共激活PI3K/AKT和ERK通路促进肿瘤细胞增殖,并促进中性粒细胞在肿瘤中的浸润,进一步促进炎症发生。此外,在肿瘤环境以及炎症环境下,白细胞介素8(IL-8)分泌增加,研究表明IL-8 可以促进肿瘤的转移以及上皮细胞-间充质转化(epithelial mesenchymal transformation,EMT)[42],在肝细胞癌中IL-8可以通过PI3K/AKT通路上调整合素β3促进肝细胞癌的进展[43]。另外有研究显示,白细胞介素37(IL-37)可以通过抑制PI3K/AKT/mTOR通路调节肝癌细胞的自噬[44]。
3 靶向PI3K/AKT/mTOR信号通路的药物研究与应用
3.1 PI3K抑制剂的研究及应用
第一代PI3K特异性抑制剂是化合物LY294002。Ma等[45]发现该化合物作用于HCC后细胞活力显著降低、凋亡增加、迁移侵袭能力下降。进一步的机制研究发现,LY294002治疗能够下调PI3K/AKT诱导的AEG-1的表达并抑制AKT、GSK3β的磷酸化。除了作用于PI3K/AKT通路发挥作用,一项临床前研究还发现LY294002能够增加肝细胞癌对放射治疗的敏感性[46]。然而,由于单独应用第一代PI3K抑制剂显示出了较大的毒副作用,因此限制了其临床应用[47]。
阿培利西(Alpelisib)是一种新型口服PI3K抑制剂,已被FDA批准用于具有PIK3CA突变的乳腺癌治疗。在肝癌临床前模型中,阿培利西同样可以特异性地治疗具有PIK3CA突变的HCC,然而在PI3K/AKT通路激活但不存在PIK3CA突变的HCC中阿培利西并没有显示出显著的治疗效果,通过联合使用Alpelisib与mTOR或CDK4/6抑制剂可以进一步提升PIK3CA突变HCC的治疗效果[48]。
伊德拉利西(Idelalisib,GS-1101,CAL-101)是一种选择性PI3Kδ抑制剂[49],已获批用于慢性淋巴细胞白血病/小淋巴细胞性淋巴瘤的治疗。在肝癌中,伊德拉利西可抑制HCC细胞增殖并诱导半胱天冬酶依赖性细胞凋亡,机制上其诱导的细胞凋亡有赖于Bim的上调[50]。
除了单独用药,近年来基于PI3K抑制剂的联合治疗研究也显示出较好的临床应用前景。研究表明,科潘利西(Copanlisib)与索拉非尼(Sorafenib)联合用于人HCC时表现出协同作用,提示该联合治疗方案在肝癌治疗中具有潜在合理性[51]。
3.2 AKT抑制剂的研究及应用
近年来,多个研究团队对于不同的新型AKT抑制剂在肝癌治疗中的效果及作用机制行了评估。例如,Sokolowski等[52]发现,变构AKT抑制剂MK2206可通过抑制PI3K/AKT和Notch1双重途径诱导HCC细胞凋亡并引起细胞增殖停滞,但MK2206 在体内HCC中是否同样具有双重靶向的效果仍有待进一步研究。DC120是一种靶向于AKT的ATP结合位点的新型人工合成化合物,除了直接抑制AKT之外,DC120还能够以剂量依赖性和时间依赖性的方式下调FKHR、FKHRL1、GSK3β和mTOR的磷酸化水平,达到抑制肿瘤细胞生长、增殖的效果[53]。此外,一些传统中草药中也含有类似AKT抑制剂的成分。Zhuo等[54]发现臭椿酮(Ailanthone)可通过抑制PI3K(p110α)的表达和AKT的Ser位点的磷酸化,在肝癌Huh7细胞中发挥抗肿瘤活性;毛兰素(Erianin)被证实以剂量依赖性方式抑制HCC细胞的增殖、迁移和侵袭,并可诱导HCC细胞的凋亡,而其作用机制与PI3K/AKT,p38和ERK/MAPK信号通路的抑制相关[55]。
在联合用药方面,研究人员发现DC120与MEK/ERK通路抑制剂U0126联用,可通过同时抑制PI3K/AKT/mTOR和MEK/ERK通路在体内协同抑制肿瘤生长[56]。SC66 能够有效抑制总AKT和磷酸化AKT的表达,并可以剂量和时间依赖的方式抑制HCC的细胞活力和集落形成能力,SC66 与多柔比星(doxorubicin)或依维莫司(RAD001)联合治疗能够更加强力地抑制体外培养Hep3B细胞和小鼠体内移植瘤的生长[57]。ARQ092 与索拉非尼联合使用在HCC大鼠模型治疗中显示出良好的耐受性和更强的抗肿瘤作用,并且显示出了较好的抗纤维化作用,提示这种联合治疗方案可能在肝硬化基础上发生肝癌的患者中具有潜在的治疗优势[58-59]。
3.3 mTOR抑制剂的研究及应用
mTOR的异常激活在肝癌等多种肿瘤中极为常见,因此目前针对mTOR抑制剂治疗肝癌的研究也最为普遍[60]。常见的mTOR抑制剂主要分为两代。第一代mTOR抑制剂主要是雷帕霉素类似物及其衍生物,这些药物在临床前研究中均显示出了对肝癌细胞生长显著的抑制作用。例如,替西罗莫司(CCI-779)可以诱导肝癌细胞出现细胞周期G1期阻滞,以此抑制体内、外肝癌的生长[61]。在小鼠肝癌模型中,依维莫司能够以计量依赖的方式抑制肿瘤的生长[62],而长期的依维莫司治疗则能够显著延缓DNA损伤诱导的肿瘤进展[63]。鉴于在临床前研究中取得的良好效果,相关药物也在临床中开展了研究。然而,第一代mTOR抑制剂在临床试验中的表现并不尽如人意。尽管在1/2期随机研究中依维莫司均显示有一定的临床治疗效果[64],但在随后的多中心3 期临床研究中,依维莫司治疗并没有明显改善出现肿瘤进展或不能耐受索拉非尼治疗的肝癌患者的总体预后[65]。而替西罗莫司更是在1/2期临床试验中即遭遇失败[66]。
因此,为了进一步提升治疗效果,第二代mTOR抑制剂应运而生。相较于第一代mTOR抑制剂,第二代抑制剂能够同时抑制由mTORC1 和mTORC2催化的下游基因磷酸化进程。以双mTOR抑制剂MLN0128为例,该化合物能够通过同时抑制mTORC1和mTORC2限制肿瘤细胞生长并诱导细胞凋亡[67],在AKT/c-MET小鼠肝癌模型中,单独给予口服MLN0128治疗也可以在一定程度上抑制肿瘤的生长[68]。另一个双mTOR抑制剂CC-233(ATG-008)在体外肝癌细胞系、人原代肝癌细胞以及小鼠移植瘤模型中均显示出了强力的杀伤作用[69]。因此,相关研究团队也对CC-233的临床应用可能进行了评估(NCT01177397),然而目前为止相关的研究结果并未公布。
在基于mTOR抑制剂的联合治疗方面,前期已开展的联合用药相关临床试验主要针对mTOR抑制剂联合索拉非尼或抗血管生成抑制剂贝伐珠单抗方案进行了研究,从目前已公布的研究结果来看,替西罗莫司联合贝伐珠单抗的方案在一项Ⅱ期临床研究中显示出了较好的应用潜力,作为一线治疗,该方案的客观缓解率达到19%,患者总体生存期为14个月[70]。然而,其余已公布联合治疗方案的效果均不尽如人意[71-72]。因此,近年来各研究团队主要针对其他潜在的联合治疗方案在临床前模型中进行了探索,以期寻找更加有效的联合治疗策略。本课题组前期研究发现,MLN0128与肝癌二线靶向药物卡博替尼联合应用可以在肝癌细胞系和部分小鼠肝癌模型中协同抑制肝癌[73]。此外,有研究显示VitD可以重新致敏对依维莫司治疗耐药的HCC细胞,机制上VitD可以通过上调miR-375 的表达,引起下游负责EMT和耐药性的几种癌基因表达的下调,从而使依维莫司治疗重新有效[74]。另有研究显示,mTOR抑制剂和苦参碱的联用能增强树突状细胞的活化和分化并显著提高体内外抗肿瘤免疫效果[75]。可见基于mTOR抑制剂的联合治疗策略可能为进展期肝癌的治疗提供新的可能,然而上述体外研究的结论仍需进一步在临床试验中进行验证。
4 小结与展望
随着肿瘤信号通路中关键靶点的不断发现,分子靶向治疗在近年来得到了飞速的发展。作为在肝癌发生与发展中最关键的信号传导通路之一,PI3K/AKT/mTOR通路成为肝癌靶向治疗的研究热点。综合近期的研究进展不难看出,一些特异性作用于通路中关键基因突变的药物在具有相应突变的肝癌亚型中具有良好的治疗效果,提示这些靶向药物在未来肝癌的精准化治疗中可能具有重要意义。然而,其余多数靶向制剂在临床应用中仍然存在毒副作用大、药物耐受等问题,此外由于肝癌异质性强以及PI3K/AKT/mTOR通路常与其他肿瘤信号通路存在串扰等原因,这些药物在临床应用中具有一定的局限性。因此,进一步探索通过联合使用PI3K/AKT/mTOR通路抑制剂和靶向其他关键通路的抑制剂在肝癌中的治疗将是未来研究的主要方向。另外,免疫检查点抑制剂联合靶向药物治疗在近年来的肝癌临床治疗中取得了重大的成果,近期的研究也发现PI3K/AKT/mTOR通路在肝癌免疫微环境的调控中同样发挥着重要的作用,包括肿瘤相关巨噬细胞极化、中性粒细胞浸润等都受到该通路的调节。因此,通过联合PI3K/AKT/mTOR通路抑制剂与肿瘤免疫检查点抑制剂治疗肝癌的相关研究也将会成为未来研究的重点。
猜你喜欢
保健医苑(2022年5期)2022-06-10
中国临床医学影像杂志(2021年6期)2021-08-14
肝博士(2020年5期)2021-01-18
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
上海农业学报(2017年3期)2017-04-10
安徽医科大学学报(2015年9期)2015-12-16
医学研究杂志(2015年7期)2015-06-22
中国当代医药(2015年16期)2015-03-01
中国药理学通报(2014年2期)2014-05-09
