
Elaidic acid leads to mitochondrial dysfunction via mitochondria-associated membranes triggers disruption of mitochondrial calcium fluxes
2024-02-16 07:14:30HuiLiuXuenanLiZiyueWangLuLiYucaiLiHaiyangYanYuanYuan
食品科学与人类健康(英文) 2024年1期
Hui Liu,Xuenan Li,Ziyue Wang,Lu Li,Yucai Li,Haiyang Yan,Yuan Yuan*
College of Food Science and Engineering,Jilin University,Changchun 130062,China
Keywords:Elaidic acid (EA)Mitochondria-associated membranes (MAMs)Calcium Endoplasmic reticulum Mitochondria dysfunction
ABSTRACT Elaidic acid (EA) stimulation can lead to endoplasmic reticulum stress (ERS),accompanied by a large release of Ca2+,and ultimately the activation of NLRP3 inflammasome in Kupffer cells (KCs).Mitochondrial instability or dysfunction may be the key stimulating factors to activate NLRP3 inflammasome,and sustained Ca2+ transfer can result in mitochondrial dysfunction.We focused on KCs to explore the damage to mitochondria by EA.After EA stimulation,cells produced an oxidative stress (OS) response with a significant increase in ROS release.Immunoprecipitation experiments and the addition of inhibitors revealed that the increase in the level of intracellular Ca2+ led to Ca2+ accumulation in the mitochondrial matrix via mitochondria-associated membranes (MAMs).This was accompanied by a significant release of mROS,loss of MMP and ATP,and a significant increase in mitochondrial permeability transition pore opening,ultimately leading to mitochondrial instability.These findings confirmed the mechanism that EA induced mitochondrial Ca2+ imbalance in KCs via MAM,ultimately leading to mitochondrial dysfunction.Meanwhile,EA induced OS and the decrease of MMP and ATP in rat liver,and significant lesions were found in liver mitochondria.Swelling of the inner mitochondrial cristae and mitochondrial vacuolization occurred,with a marked increase in lipid droplets.
1. Introduction
Trans fatty acids (TFAs) are unsaturated fatty acids that contain one or more carbon-carbon double bonds in the trans configuration[1].Most of them are generated during the manufacture of partially hydrogenated or deodorized vegetable oils.Compelling epidemiological evidence suggests that intake of TFA,especially industrial TFA,increases the risk of various diseases such as cardiovascular diseases,atherosclerosis,diabetes,and Alzheimer’s disease (AD)[2,3].However,the molecular mechanisms of TFA-related chronic diseases remain to be elucidated.Elaidic acid (EA,18:1trans-9) is the main trans isomer and has recently become a major concern in the field of food safety.EA is known to induce atherosclerosis in human umbilical vein endothelial cells[4].Other research has indicated that excessive intake of EA accelerates atherosclerosis by promoting inflammation and oxidative stress(OS) in a mouse model of hyperlipidemia[5].Consumption of EA exacerbates hyperlipidemia and fatty liver change in zebrafish[6].EA contributes to energy and lipid metabolism,and was confirmed as a potential biomarker for high fructose-induced nonalcoholic fatty liver disease (NAFLD)[7].Studies have shown that EA causes apoptosis in a variety of cells[8-10],increased endoplasmic reticulum stress (ERS)and OS in diabetic mice[11],and activated NLRP3 inflammasome in macrophages[12],but its specific pathogenic mechanism in chronic diseases remains to be further studied.Previous studies have shown that EA induces ERS in Kupffer cells (KCs),accompanied by a concentration increase in intracellular Ca2+,leading to the formation of the NLRP3 inflammasome and the release of inflammatory factors[13].Blocking mitophagy leads to the accumulation of damaged mitochondria,increasing the activation of the NLRP3 inflammasome,which links mitochondrial damage to NLRP3 inflammasome activation[14].It is worth noting that maternal intake of TFA during pregnancy and lactation can impair mitochondrial bioenergetics in the liver[15].Therefore,the role of EA on mitochondria remains to be further investigated.
It has been shown that many stimuli make Ca2+signaling an intermediate step in triggering mitochondrial destabilization,generating mitochondria-associated ligands that activate the NLRP3 inflammasome[16].Ca2+mobilization leading to Ca2+overload and/or mitochondrial damage can trigger NLRP3 inflammasome activation,and the Mitochondria-Associated Membranes (MAM) that support this model are required for NLRP3 inflammasome activation,which is essential for Ca2+transfer from the ER to the mitochondria[17].Mitochondrial dysfunction has been observed in many settings of NLRP3 inflammasome activation,and inhibition of Ca2+mobilization reduces mitochondrial reactive oxygen species (mROS) production[18].Mitochondria take up Ca2+during NLRP3 inflammasome activation via membrane attack complexes and shRNA knockdown of the mitochondrial calcium uniporter (MCU)[19],which decreases mitochondrial Ca2+uptake and reduces IL-1β production[20].
In particular,the ER transmits appropriate Ca2+signals to the mitochondria,which decodes them into specific signals to regulate essential functions,including metabolism,energy production,and apoptosis.Mitochondria are not only the energy generators of the cell,but also the main hub of cellular Ca2+signaling,which is essential for cell survival and death[21,22].Mitochondria are crucial to cell fate,in large part because they are involved in the dynamic regulation of cellular Ca2+.Under physiological conditions,Ca2+accumulation in mitochondria stimulates oxidative metabolism through the regulation of Ca2+sensitive dehydrogenases and metabolite carriers[23].Ca2+is transferred from the ER to the mitochondria through a tight contact between the two organelles.These connections are known as MAM,and play a key role in maintaining intracellular Ca2+homeostasis[24].The main role of inositol 1,4,5-trisphosphate receptors (IP3R) located in ER is to allow the transfer of Ca2+from the ER to intracellular stores,particularly in the mitochondria.The voltage-dependent anion channel (VDAC) is a channel located in the outer mitochondrial membrane (OMM),and molecular chaperone protein glucose regulatory protein 75 (Grp75) is a bridging protein that physically interacts with IP3R and VDAC1.Knockdown of Grp75 disrupts this complex and reduces mitochondrial Ca2+uptake[25].
Mitochondrial Ca2+overload has long been known to be a critical event in the bioenergetic crisis associated with cell death by necrosis and acts as a critical sensitizing signal in the intrinsic apoptosis pathways[26].Mitochondrial Ca2+uptake promotes oxidative phosphorylation and mROS production,but excessive and/or sustained Ca2+influx leads to mitochondrial Ca2+overload and decrease of adenosine triphosphate (ATP) production[27-29].Ca2+and reactive oxygen species (ROS) are the most important triggers for mitochondrial permeability transition pore (MPTP) opening,acting in living cells in conjunction with a variety of pathological challenges[30,31].Irreversible MPT dissipates mitochondrial membrane potential (MMP),disrupts oxidative phosphorylation,promotes mROS production,and may lead to osmotic swelling and mitochondrial rupture.Thus,mROS,Ca2+and MPT form part of an amplification circuit that triggers irreversible mitochondrial damage[30],which leads to disruption of the mitochondrial mass control system,Ca2+dysregulation and involvement in autophagy,ER stress,inflammation and apoptosis[32,33].Furthermore,mitochondrial Ca2+levels are more sensitive to changes in ER Ca2+kinetics than to overall Ca2+signaling[34],emphasizing the role of MAM-mediated Ca2+communication between ER and mitochondria in maintaining mitochondrial function and intracellular homeostasis.
The Ca2+transfer at MAM may be of great significance,and this process is closely related to the induction of mitochondrial damage,including the production of mROS and MPT induction.A growing number of publications have described the molecular composition and possible involvement of MAM in pathology,highlighting the importance of these interactions in cellular physiology and pathology[35].However,its specific mechanism of action still needs to be studied in depth.Currently,little is known about the role of MAM-mediated Ca2+communication in TFA-induced inflammatory responses.This paper focused on the mechanisms by which EA induced Ca2+disruption may ultimately lead to mitochondrial damage and the relevance of ER-mitochondrial interactionsinvivo.In addition,in vitroexperiments were conducted to verify whether EA caused damage to liver mitochondria in SD rats.This research provides theoretical support for the reduction of TFA damage in the organism.
2. Materials and methods
2.1 Chemicals and reagents
EA (C18:1T,CAS 112-79-8) was purchased from ANPEL Laboratory Technologies Co.,Ltd.(Shanghai,China).2’,7’-Dichlorofluorescein diacetate (DCFH-DA),JC-1 staining probe,BCA protein assay kit,enhanced ATP assay kit,MPTP assay kit,DAPI staining probe and immunol fluorescence staining kit were obtained from Beyotime Biotechnology (Shanghai,China).Rat ROS assay kit was purchased from Shanghai Youxuan Biotechnology Co.,Ltd.(Shanghai,China).Fluo-4 AM staining probe was purchased from Dojindo Laboratories (Japan).Rhod-2 AM was bought from Abcam (Cambridgeshire,UK).The enzymatic assay kits for detecting superoxide dismutase (SOD),glutathione (GSH) and malondialdehyde(MDA) were purchased from Nanjing Jiancheng Bioengineering Institute(Nanjing,China).The MitoSOX Red mitochondrial superoxide indicator was bought from Thermo Fisher (Waltham,MA,USA).
2.2 Cell culture and treatment
KCs (BeNa Culture Collection,BNCC,Beijing,China) were cultured in 1640 complete medium,supplemented with 89% RPMI 1640 medium (Gibco BRL Co.,Ltd.,Grand Island,NY,USA),10%(V/V) fetal bovine serum (Gemini Bio-products,Woodland,USA),0.5%L-glutamine (Beijing Dingguo Changsheng Biotechnology Co.,Ltd.,Beijing,China),as well as 0.5% penicillin and streptomycin(Hyclone Corporation,Logan,UT,USA).KCs were revived in 10 cm sterile cell culture dishes (Corning Incorporated,New York,USA),and then incubated at 37 °C for 24 h with 5% CO2.All these reagents were diluted to different concentrations using serum-free RPMI 1640 medium and filtered through a 0.22 μm membrane(Millex-GP,Merck Millipore Ltd.,Tullagreen Carrigtwohill,Co.,Cork,IRL).
2.3 Animals and experimental design of EA exposure
The male Sprague Dawley (SD) rats,aged 8 weeks and weighing 200 g,were purchased from Liaoning Changsheng Biotechnology Co.,Ltd.The experiments were implemented in conformity to the Guideline for Animal Experimentation of Jilin University (Changchun,China).The experimental procedures were approved by the Ethical Welfare Committee of Jilin University (No.SY202105011).After adaptive feeding for one week,36 male rats were randomly classified into 4 groups: (i) a negative control group which was given gavage corn oil with 1.0 mL/100 g bw daily;(ii) EA-exposed groups that received gavage with 50,100 and 150 mg/kg bw EA in corn oil.The experiment was conducted for 4 weeks.The dosage was determined based on pre-experiment and literature references[5,12,36].All rats were housed 3/cage under constant environmental conditions (12 h light/dark cycle,20−25 °C,(55 ± 5)% humidity) and had free access to standard feed and drinking water.The body weight was recorded once a week.The SD rats were later anesthetized with sodium pentobarbital and blood was taken from the heart.SD rats were quickly dissected and the organs were taken out and frozen at −80 °C.
2.4 Measurement of OS indicators in KCs and rat liver
SOD,GSH and MDA activities in cells and liver tissues were assayed using commercial kits according to the manufacturer’s instructions.Protein levels were measured with a BCA protein assay kit.
2.5 Measurement of intracellular ROS,mROS and rat liver ROS
Cellular ROS levels were determined by a fluorescent probe(DCFH-DA).KCs were seeded at a density of 1 × 105cells/well in 6-well microtiter plates.After 24 h of adnate growth,KCs were treated with different concentrations of EA (0,0.05,0.1 mmol/L) for 6 h.KCs were washed three times with PBS (Hyclone Corporation,Logan,UT,USA),followed by the addition of 10 μmol/L DCFH-DA and incubation at 37 °C for 30 min in the dark.Then,KCs were washed three times with PBS and the fluorescent intensity was detected using a fluorescence microscope (Nikon TS100,Japan).Liver tissue ROS assays were based on rat ROS assay kit.Rat liver homogenate was prepared according to the kit instructions for determination.
Mitochondrial superoxide is the predominant ROS in mitochondria.The mROS level was detected by the MitoSOX Red mitochondrial superoxide indicator.Cells were incubated with green mitochondrial fluorescent probe working solution for 30 min at 37 °C,followed by MitoSOX solution (5.0 μmol/L) for 30 min at 37 °C,and were then stained with DAPI for 4 min at room temperature protected from light,and quantified using a spectral laser confocal microscope(FV-1000,Olympus,Japan) to detect the MitoSOX.
2.6 Measurement of cytosolic Ca2+ and mitochondrial Ca2+ in KCs
Cytosolic Ca2+levels were determined using Fluo-4 AM.Cells were loaded with 4 µmol/L Fluo-4 AM for 30 min at 37 °C and then washed and maintained in PBS for testing by Flow Cytometer(FACSCalibur,Becton,Dickinson and Company,USA).Rhod-2 AM was used to measure the mitochondrial Ca2+level.Cells were incubated with 4 µmol/L Rhod-2 AM for 30 min at 37 °C,and evaluated by flow cytometry.To investigate whether the EA-induced increase in cellular mitochondrial Ca2+was partly due to the transfer from the ER to the mitochondrial via the MAM,cells were stimulated with the IP3R inhibitor 2-aminoethoxydiphenyl borate (2-APB,10 μmol/L,ApexBio Technology,USA) and the MCU inhibitor RU360 (10 μmol/L,Merck,USA).Changes in cytoplasmic Ca2+and intra-mitochondrial Ca2+were measured,respectively.
2.7 Transmission electron microscopy (TEM) of KCs and rat liver tissue
The organelle morphology and the structure of cells and tissues were observed under the transmission electron microscope.Briefly,crude mitochondrial fraction samples were fixed with 2.5% glutaraldehyde,post-fixed in osmic acid solution,dehydrated with a graded alcohol series and acetone,and finally embedded in embedding media.Ultrathin sections were stained with uranyl acetate and lead citrate,and examined under a JEM 1400 (JEOL Ltd.,Japan)transmission electron microscope operated at 80 kV.
2.8 MMP,ATP and MPTP measurement in KCs and rat liver
JC-1 (Beyotime Biotechnology,Shanghai,China) was used for detecting MMP (Δψm) of fluorescent dyes in cells by flow cytometry and the level of MMP in rat liver was detected by a fluorescence spectrophotometer (RF-5301PC,SHIMADZU,Japan).ATP contents in cells and liver tissues were measured by an enhanced ATP assay kit following the manufacturer’s instructions.A fluorescent probe for membrane permeability,Calcein AM,was used to detect the degree of MPTP opening in cells,which was finally detected by Flow Cytometer.
2.9 Double staining of mitochondria and ER in KCs
Cells and mitochondria-ER staining working solution (1 μmol/L ER-Tracker Red and 1 μmol/L Mito-Tracker Green dye) (Beyotime Biotechnology,Shanghai,China) were incubated in the dark for 30 min at 37 °C.After staining,the cells were washed 3 times with serum-free medium and then the mitochondrial and ER colocalization was observed by spectral laser confocal microscope.
2.10 Immunocytochemical staining for protein colocalization
The degree of protein co-localization was observed by immunofluorescence.Cells were rinsed twice in PBS,prior to fixation with 4% formaldehyde for 30 min,and then fixed with fixative for 60 min.Cells were labeled with the target primary antibody overnight at 4 °C and then incubated with the specific fluorescent secondary antibody for 1 h at room temperature in a shaker and finally incubated with DAPI for 4 min,referring to the instructions of the immunofluorescence staining kit for details.Cells were imaged on a spectral laser confocal microscope.
2.11 Western blotting analysis
After EA treatment,cells were washed three times with precooled PBS and lysed on ice with RIPA lysis buffer containing 1% protease and 1% phosphatase inhibitor cocktails (Beyotime Biotechnology,Shanghai,China) for 30 min.The lysates were collected and centrifuged at 10 010 ×gfor 10 min at 4 °C.BCA protein assay kit was used to quantify the protein concentrations of each sample,and then proteins were denatured in loading buffer(Beyotime Biotechnology,Shanghai,China) at 100 °C for 10 min.
Equal amounts of protein were resolved on 10% or 12% SDS-PAGE gels.The target proteins were identified using Rainbow Maker(Thermo Fisher Scientific,USA).After electrophoresis,the proteins were transferred onto polyvinylidene fluoride membranes (PVDF,Millipore,Billerica,MA,USA) by semi-dry transmembrane instrument (Bio-Rad Laboratories,California,USA).The PVDF membranes were blocked with a blocking solution (Beyotime Biotechnology,Shanghai,China) at room temperature for 1 h.After blocking,the membranes were incubated with corresponding specific antibodies overnight at 4 °C.The membrane was washed and incubated with IgG secondary antibodies for 2 h at room temperature.The target protein reacted with enhanced chemiluminescence (ECL)chromogenic substrate (Beyotime Biotechnology,Shanghai,China).The images were detected using Scan LiDE 100 scanner (Canon,Japan) and the gray values were analyzed using Image-J software.
For western blot analysis,rabbit polyclonal anti-IP3R (ab108517,Abcam,UK),rabbit monoclonal anti-Grp75 (14887-1-AP,Proteintech Group,Inc,USA),mouse monoclonal anti-VDAC1 (66345-1-Ig,Proteintech Group,Inc,USA),rabbit monoclonal anti-MCU (D2Z3B,#14997,Cell Signaling Technology,USA),and rabbit monoclonal anti-GAPDH (ab181602,Abcam,UK),were used.
2.12 Immunoprecipitation (IP) analysis of the IP3R-Grp75-VDAC1 complex
Cells were collected using ice-cold IP lysis buffer containing 1% phosphatase inhibitor cocktails for 10 min.The lysates were transferred to a micro centrifuge tube and centrifuged at 14 000 ×gfor 10 min to pellet the cell debris.Supernatant of 50 mL was aspirated as input group,and the remaining protein was used for subsequent experiments.The protein samples were bound to the antibody binding flip incubation overnight at 4 °C,and then bound to the washed Protein A/G Magnetic Beads (MedChemExpress LLC,USA) at room temperature for 1 h.Then 50 μL of 1× SDS-PAGE Loading Buffer was added to the beads,mixed well and heated at 100 ºC for 5 min.Beads were then removed,and the supernatant was collected and SDS-PAGE assay was conducted.Western blotting and semi-quantitation were performed as the method described above.
2.13 Statistical analysis
Statistical analysis and graphs were performed using GraphPad Prism 7.0 software (GraphPad Software,Inc.,La Jolla,CA,USA).The data were expressed as the mean ± standard deviation (SD),analyzed by ANOVA and the Newman-Keuls multiple-comparison test.Difference between the mean values was considered significant atP<0.05.
3. Results
3.1 EA promoted OS in KCs
ROS act as signaling molecules in cellular biological processes and this is the basis for normal cellular function,but excessive ROS can lead to OS.The fluorescence intensity of ROS showed that EA significantly increased the release of ROS (P<0.01,Fig.1A).OS can be directly assessed by measuring ROS release.Our study indicated that EA treatment induced imbalance of redox state in KCs,further leading to OS.Compared with the control group,the enzymatic activities of SOD(P<0.01) and GSH (P<0.01) were significantly reduced,and MDA values (P<0.05) increased significantly with increasing doses of EA (Fig.1B),indicating that EA induced OS in KCs.
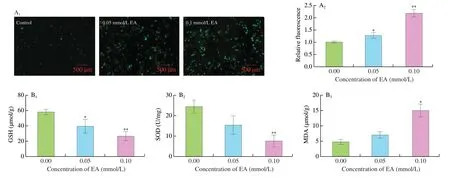
Fig.1 Effects of EA on OS in KCs.(A) Effects of EA on ROS release measured by fluorescence microscope in KCs.(B) Effect of EA on SOD,GSH and MDA in KCs.Values are presented as means ± SD (n=3).Significant differences with the control group were designated as * P <0.05 or ** P <0.01.
3.2 Mitochondria-associated ER-membranes and Ca2+regulatory axis
Results demonstrated that the co-localization of mitochondria with ER was significantly enhanced in EA-induced cells compared with the control group (Fig.2A).IP3R is a signature protein of ER and VDAC1 is a signature protein of mitochondria.EA induced the co-localization between IP3R and VDAC1 in cells as observed by laser confocal microscopy (Fig.2B).We also detected the interaction of IP3R,Grp75,and VDAC1 by immunoprecipitation,and found that the interactions between IP3R and Grp75,Grp75 and VDAC1 were significantly enhanced,and that protein expression was significantly changed in the experimental group compared to the control (P<0.01,Fig.2C).This indicated that EA induced the formation of the cellular IP3R-Grp75-VDAC1 complex and enhanced the MAM-coupled structure.

Fig.2 Mitochondria-associated ER-membranes and Ca2+ regulatory axis.(A) Immunofluorescence analysis of mitochondrial (green) and ER (red) compartments of KCs.Individual channels and color-merged image are presented and the contact points between the two organelles are indicated with a white arrow.(B) The colocalization of IP3R and VDAC1 was determined by immunofluorescence.Blue: nuclear staining;Green: fluorescent secondary antibody corresponding to IP3R;Red: fluorescent secondary antibody corresponding to VDAC1.The contact point between the mitochondrial and ER is the bright yellow area indicated by the white arrow.(C) IP was performed to analyze the IP3R-Grp75-VDAC1 interaction.(D) Effect of EA on mitochondria Ca2+ in KCs.(E) Effect of IP3R inhibitor 2-APB on cytoplasmic Ca2+ in KCs induced by EA.(F) Effect of MCU inhibitor RU360 on mitochondria Ca2+ in KCs induced by EA.(G) Effect of 2-APB and RU360 on MCU protein expression in KCs induced by EA.* P <0.05 and ** P <0.01,versus the control group;# P <0.05 and ## P <0.01,versus the 0.1 mmol/L EA-treated group.
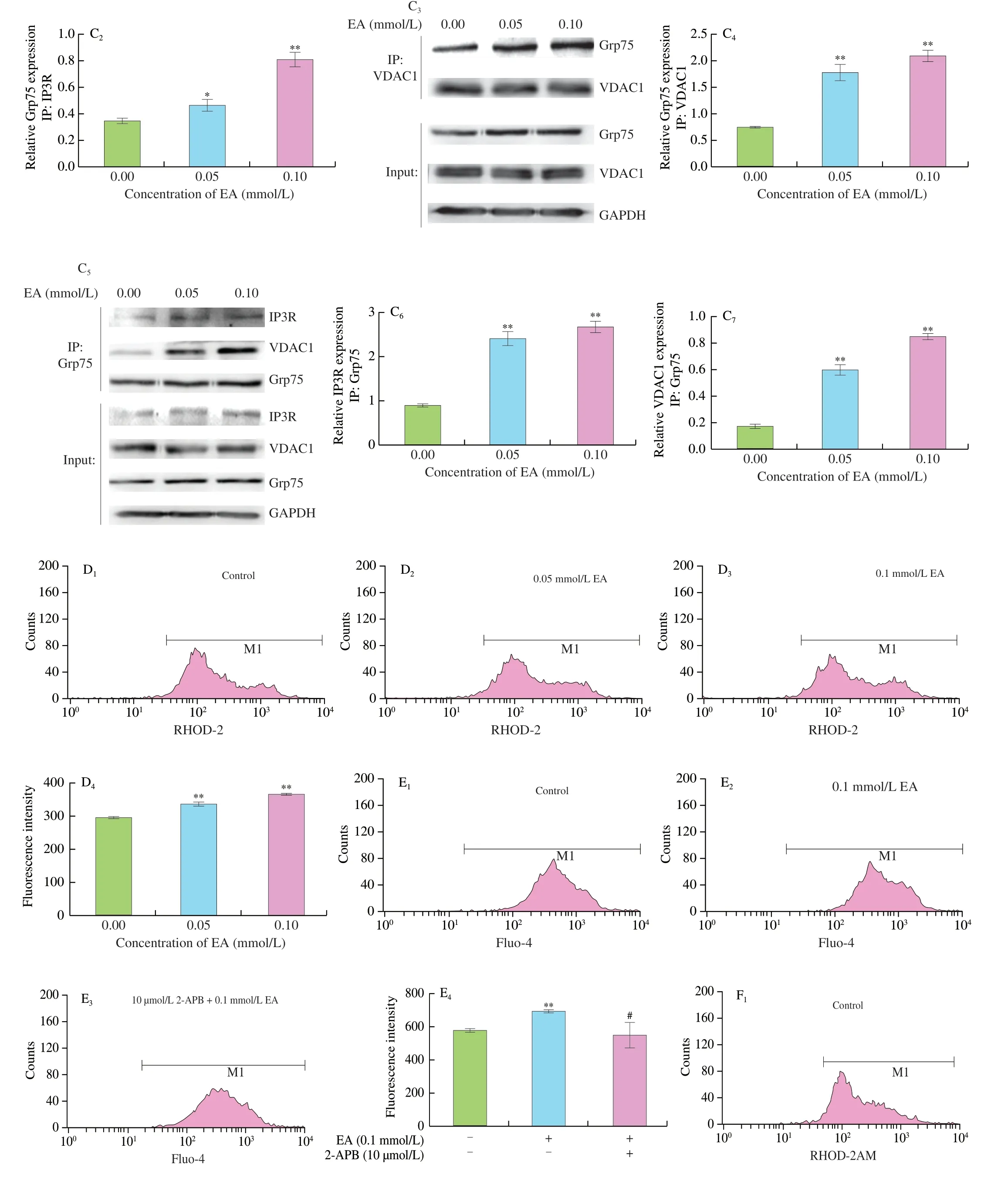
Fig.2 (Continued)
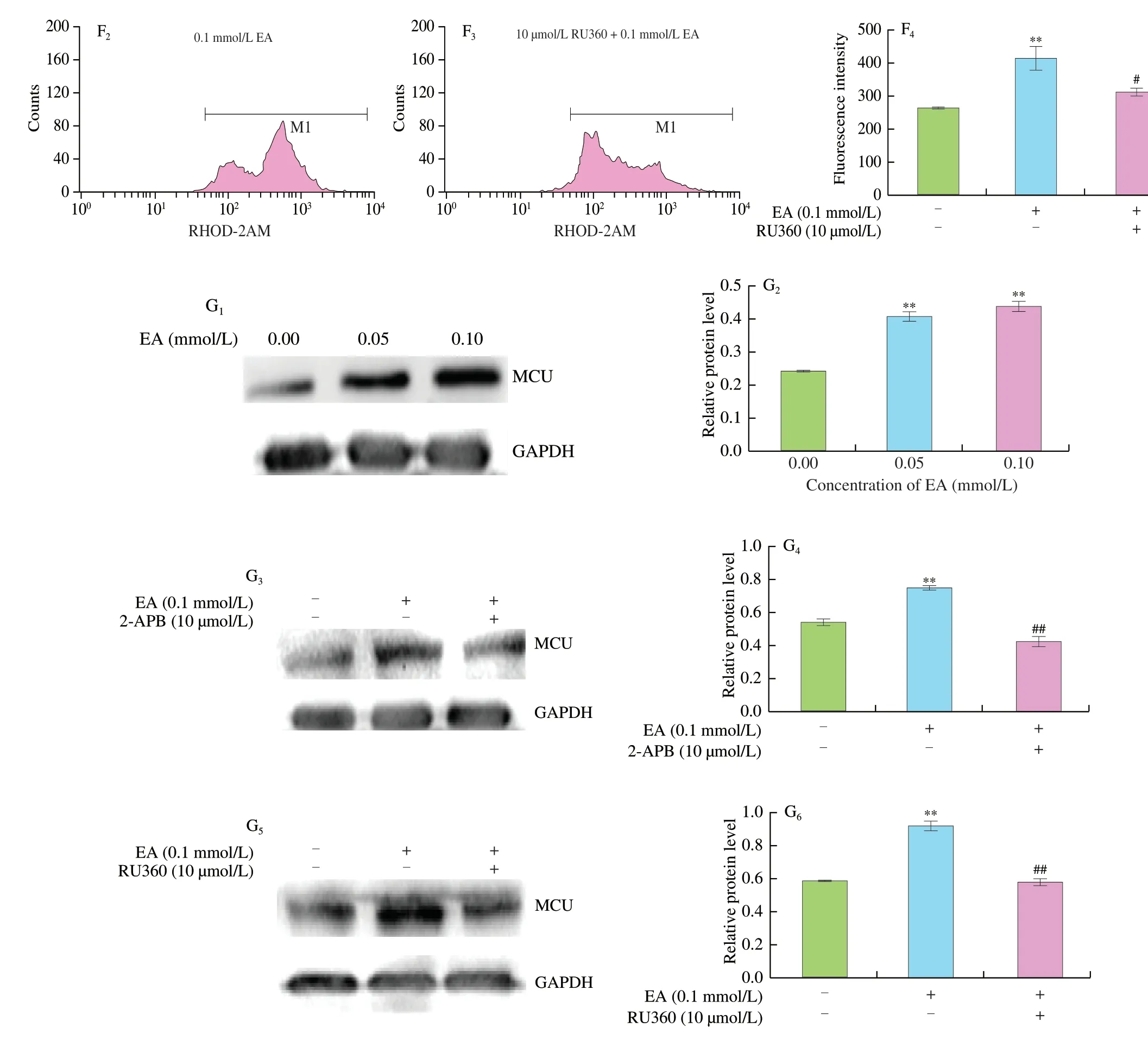
Fig.2 (Continued)
Flow cytometry results showed that EA induced an increase in mitochondrial Ca2+levels (P<0.01,Fig.2D).To further confirm that the EA-induced cytoplasmic and mitochondrial Ca2+upregulation originated from the MAM,2-APB was used to inhibit Ca2+release from the ER.The results showed that 2-APB significantly decreased the EA-induced elevation of cytoplasmic Ca2+levels (P<0.05,Fig.2E),while the MCU inhibitor RU360 significantly inhibited the increase of mitochondrial Ca2+levels (P<0.05,Fig.2F).The expression of MCU protein increased significantly with the increase of EA concentrations.In contrast,the addition of both inhibitors significantly decreased MCU expression (P<0.01,Fig.2G).The above results suggest that EA enhanced the MAM structure and facilitated Ca2+transport from the ER to mitochondria.
3.3 EA promoted mitochondrial dysfunction in KCs
EA induced the marked release of ROS.Mitochondria are considered to be the main source of ROS and are also particularly vulnerable to oxidative damage.Confocal laser scanning results showed a significant increase in MitoSOX fluorescence intensity,demonstrating an increase in mROS release (Fig.3A).It was shown that the opening of the MPTP could increase the permeability of the inner mitochondrial membrane,disrupt the membrane potential,cause uncoupling of oxidative phosphorylation,and prevent mitochondria from producing ATP through oxidative phosphorylation,which is the main source of intracellular synthesis of ATP.Decreased ATP levels are associated with impaired mitochondrial function and can affect normal cellular physiological functions.Flow cytometry values representing the green fluorescence intensity of calcein are used to determine the degree of MPTP opening.These values gradually decreased with increased EA concentration,demonstrating a decrease in green fluorescence and a significant increase in mPTP opening(P<0.01,Fig.3B).The ATP level also decreased significantly(P<0.01,Fig.3C).A stable MMP is a prerequisite for maintaining mitochondria for oxidative phosphorylation and for maintaining normal cellular function.After EA stimulation,the MMP was significantly downregulated (P<0.01,Fig.3D).

Fig.3 Effect of EA on mitochondrial dysfunction in KCs.(A) Effect of EA on mROS detected by laser confocal microscopy;Blue: DAPI;Green: mitotrackerTM Green FM;Red: mtSOX Deep Red.(B) The opening of MPTP induced by EA was detected by flow cytometry.(C) Effect of EA on ATP concentration was determined by fluorescent microplate reader.(D) Effect of EA on MMP was examined by flow cytometry.Significant differences with the control group were designated as * P <0.05 or ** P <0.01.
3.4 Morphological and structural changes of mitochondria in KCs
The effect of EA on the mitochondrial morphology of KCs was further investigated using TEM (Fig.4).The inner mitochondrial ridge was an important indicator of mitochondrial function,and the inner ridge of mitochondria in the control group was smooth and neatly arranged.After EA induction,cell mitochondria showed mitochondrial damage such as swelling of the inner ridge and mitochondrial vacuolization,suggesting that EA caused structural changes in cell mitochondrial morphology.The graph visually revealed a significant increase in the number of MAM,the coupling structure between mitochondria and ER.
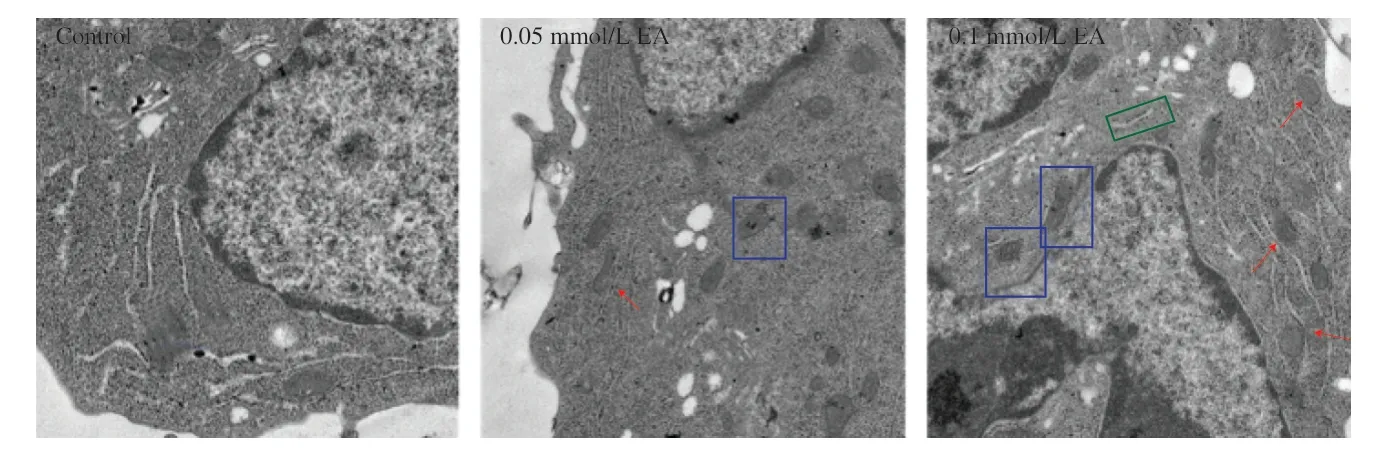
Fig.4 Effect of EA on cell ultrastructure (at ×30 000 magnification).The red arrow represents MAM,the blue circle represents mitochondria,and the green box represents ER.
3.5 EA-induced OS and mitochondrial damage in rats
As seen in Fig.5A,compared with the control group,the release of ROS from the liver of the model group was significantly increased,while the enzymatic activities of SOD and GSH were significantly decreased and the MDA values were significantly increased with the increase of EA demonstrating the occurrence of EA-induced OS injury in the liver of SD rats (P<0.01).

Fig.5 Effects of EA on OS and mitochondria damage of SD rat liver.(A) Effects of EA on ROS release and SOD,GSH,MDA concentrations in SD rat liver.(B) Effect of EA on ultrastructure of rat liver (at 25 000× magnification).The red arrow represents MAM,the blue circle represents mitochondria,and the green box represents ER.(C) ATP concentration was determined by fluorescent microplate reader,MMP was examined by fluorescence spectrophotometer.Significant differences with the control group were designated as * P <0.05 or ** P <0.01.
TEM examination of the liver revealed that the nuclear membrane of the control group was round or oval,with an intact nuclear membrane.The morphology of mitochondria was normal,the cristae column structure was clear,the structure of rough ER was normal,the ribosomes were rich and there were few lipid droplets.Conversely,in the model group,the nuclear membrane was irregular or blurred,and the number of lipid droplets increased significantly.The mitochondria were swollen and distorted,the cristae structure was blurred,and the rough ER was swollen,broken and disorganized (Fig.5B).These observations strongly suggested that EA caused significant damage to the morphological structure of rat liver mitochondria.It was also accompanied by a significant reduction of liver MMP and ATP with the increase of EA concentration (P<0.01,Fig.5C).
4. Discussion
Recent studies have shown that the intake of TFA is strongly associated with chronic diseases,including coronary heart disease,diabetes,and atherosclerosis[37-39].EA is the major isomer of TFA,comprising typically 20%−30% of total C18:1transisomers,and therefore serves as the main target of our study[40].EA-induced chronic diseases,such as atherosclerosis,a metabolic and inflammatory disease,are associated with a significant increase in fatty liver/liver inflammation,and the liver is a central site of inflammation and lipid metabolism,critical to atherosclerosis[41,42].Rat livers and KCs were therefore selected as the target for EA induction.
Morinaga et al.[11]and Cassagno et al.[36]reported that TFA caused OS and ERS in mouse hepatocytes and liver tissue.The onset of OS is closely linked to changes in the mitochondrial respiratory chain.Our study showed that EA induced KCs to release large amounts of ROS;the enzymatic activities of SOD and GSH were significantly reduced;and MDA values increased significantly with the increase of EA,demonstrating that EA induced OS in KCs (Fig.1).OS also occurred in SD rat liver (Fig.5A).
High levels of ROS are produced in the ER,and ROS mainly act on the Ca2+channel receptor on the MAM,resulting in extensive release of ER Ca2+into the mitochondria.During ischemiareperfusion,excess mitochondrial Ca2+leading to excessive activation of the electron transport chain was directly associated with increased ROS production[43].On the other hand,due to the new steroids,high levels of Ca2+promote mitochondrial oxidative disorders,which can lead to OS[44].EA induced ERS in KCs,accompanied by the production of large amounts of Ca2+[13].Excessive Ca2+uptake in the mitochondrial matrix can lead to mitochondrial Ca2+overload.Mitochondrial Ca2+were found to increase significantly after EA stimulation (Fig.2D).Under physiological and pathological conditions,the ER and mitochondria play a central role in Ca2+signaling,and the MAM is a specialized microregion for Ca2+translocation.The important role of MAM in maintaining mitochondrial Ca2+homeostasis is well demonstrated by the fact that MAMs mediate Ca2+translocation and regulate related proteins and their functions.Therefore,it is reasonable that a moderate and well-timed increase in Ca2+levels in the mitochondrial matrix due to enhanced MAM communication could help mitochondria adapt to stressful conditions that require more metabolic output.However,sustained Ca2+transfer can lead to mitochondrial dysfunction.
The MAM provides an excellent platform for numerous signaling pathways,and the MAM-associated inflammatory signaling pathway plays a key role in cellular defense during pathogenic infections and metabolic disorders.Many studies have demonstrated the importance of MAM in maintaining the normal function of both organelles.Abnormal numbers,structure,or function of MAM are associated with the development of inflammatory responses.Arruda et al.reported that liver MAM levels and the expression of Ca2+flux proteins associated with MAM were increased in obese mice,and that increased accumulation of Ca2+in mitochondria leads to mitochondrial dysfunction,increased ROS production,cellular stress,impaired insulin action in the liver,and abnormal glucose metabolism[45].Studies have shown that diabetes promotes the formation of MAM in podocytes,leading to sustained mitochondrial Ca2+overload in podocytes,which in turn induce mitochondrial dysfunction and the development of glomerular injury[46].Plus,Ca2+regulation axis antagonists reduce Ca2+transfer through MAM and prevent mitochondrial Ca2+overload and subsequent podocyte apoptosis in an Adriamycin nephropathy rat model[47].
The results of analysis using laser confocal microscopy (Fig.2A)revealed a significant enhancement of the MAM structure accompanied by changes in mitochondrial morphology and chromatin aggregation in the nucleus,which was consistent with the results of TEM (Fig.4).MAM transported Ca2+between ER and mitochondria via the regulatory protein IP3R and molecular chaperone protein Grp75,which are essential components of MAM.VDAC in the OMM is physically linked to the IP3R via Grp75 to form the VDAC1/Grp75/IP3R channel complex[25]and serves as a channel for signaling molecules,including Ca2+,apoptotic signals and metabolites[48].The potential for EAinduced increase in cytoplasmic Ca2+partly via IP3R channels was also investigated.The results showed that 2-APB inhibitors reduced the EA-induced increase in cytoplasmic Ca2+(Fig.2E),suggesting that Ca2+was released from the ER lumen through IP3R after MAM formation.In response to cellular/stress stimuli,Ca2+was released from the ER-mitochondrial interface and translocated through the mitochondrial membrane gap,and then eventually entered the mitochondrial matrix via MCU[49].Cardiomyocytes specifically deficient in MCU inhibited MPTP opening[50].Higher MCU activity triggered NLRP3 inflammasome activation and Caspase-dependent apoptosis[20,51].MCU inhibitor RU360 also stimulated the reduction of mitochondrial Ca2+(Fig.2F).Both 2-APB and RU360 inhibited the expression of MCU protein,respectively (Fig.2G).This indicated that the increased Ca2+induced by MAM accumulated into the mitochondrial matrix through MCU,and suggested that the increase of Ca2+concentration in mitochondria caused by EA might be related to the enhancement of MAM structure.Meanwhile,the results of immunofluorescence showed that the degree of co-localization of IP3R and VDAC1 gradually increased with the increase of EA concentrations (Fig.2B).The results of immunoprecipitation showed that the interaction between Grp75 and IP3R and VDAC1 were enhanced,respectively (Fig.2C).These findings also indicated the presence of “IP3R-Grp75-VDAC1-MCU” as an intracellular Ca2+regulatory axis based on the Ca2+transfer pathway[47].
Fig.3B shows that the opening of the MPTP increased with EA levels.It is known that excessive production of ROS can also directly induces MPTP opening[52].In addition,MPT can also be triggered by high/sustained mROS production[53].Sustained Ca2+transfer can lead to mitochondrial dysfunction,as evidenced by increased mROS production,decreased MMP,and reduced basal oxygen consumption[45].Laser confocal microscopy results revealed a significant increase in mROS after EA stimulation (Fig.3A).Elevated mROS production was suggested to be the relevant signal indicative of mitochondrial damage,as disruption of the electron transport chain was sufficient to activate the NLRP3 inflammasome[14],corresponding to our previous findings.Predictably,the yield of mROS decreased rapidly with the addition of 2-APB or RU360 (Fig.3A).The results of mitochondrial related indicators showed that ATP and MMP decreased significantly with the increase of EA concentrations(Figs.3C,D).The same occurred in SD rat liver,where the changes in mitochondrial morphology were accompanied by an increase in the amount of MAM and a significant decrease in MMP and ATP as EA concentrations increased (Fig.5C).The opening of MPTP induced mitochondrial swelling.Meanwhile,After EA stimulation,cells showed mitochondrial damage,such as swelling of inner ridge and vacuolation of mitochondria,indicating that EA caused changes in the morphological structure of cell mitochondria.In addition,it was clearly observed that the ER was more closely linked to the mitochondria,and the number of MAM increased with increasing EA concentrations.
We also observed that following EA stimulation,both cells and rat livers developed an OS response with a significant increase in ROS release.The increase in the cytosolic Ca2+concentration was achieved by the release of IP3R channels from the ER lumen.The increase in the intracellular Ca2+concentration led to Ca2+accumulation in the mitochondrial matrix via the “IP3R-Grp75-VDAC1-MCU”,the release of large amounts of mROS,and loss of MMP and ATP,accompanied by a significant increase in MPTP permeability,ultimately leading to mitochondrial destabilization.These findings confirmed the mechanism by which EA induced mitochondrial Ca2+imbalance through MAM,ultimately leading to mitochondrial dysfunction,and provided directions for future research.Upon disruption of MAM integrity,miscommunication directly or indirectly caused imbalances in Ca2+homeostasis and increased ERS and OS,contribute to the pathogenesis of kidney disease[54],and targeting MAM may be a viable strategy for NAFLD treatment[55].Control of mitochondrial Ca2+levels and precise regulation of MAM formation and function may be a promising way to inhibit the development of cardiovascular disease[56].Tian et al.balanced Ca2+homeostasis injured by excess lipids through MAM with sulforaphane[57].Further understanding of the pathophysiological mechanisms underlying these diseases will facilitate the development of new pharmacological treatments.
In summary,EA disrupted mitochondrial Ca2+homeostasis,resulting in mitochondrial destabilization,which is inextricably linked to the enhancement of MAM.In vitroexperiments also confirmed that EA caused damage to mitochondria in SD rat liver,which resulted in irregular nucleus,irregular or fuzzy nuclear membrane,swelling and deformation of mitochondria and increase of lipid droplets.
Conflict of interest
The authors declare that there is no conflict of interests.
Acknowledgments
This work was supported by fund from the National Natural Science Foundation of China (32172322).The authors gratefully acknowledge the fund support.

- 食品科学与人类健康(英文)的其它文章
- Modifications in aroma characteristics of ‘Merlot’ dry red wines aged in American,French and Slovakian oak barrels with different toasting degrees
- Effect of different drying methods on the amino acids,α-dicarbonyls and volatile compounds of rape bee pollen
- Dynamic changes in physicochemical property,biogenic amines content and microbial diversity during the fermentation of Sanchuan ham
- A comparison study on structure-function relationship of polysaccharides obtained from sea buckthorn berries using different methods:antioxidant and bile acid-binding capacity
- Yolk free egg substitute improves the serum phospholipid profile of mice with metabolic syndrome based on lipidomic analysis
- Underlying anti-hypertensive mechanism of the Mizuhopecten yessoensis derived peptide NCW in spontaneously hypertensive rats via widely targeted kidney metabolomics