
右美托咪定通过激活AMPK减轻TNF-α诱导人成神经细胞瘤细胞系SH-SY5Y损伤
2024-02-06 04:13:06杨志文
基础医学与临床 2024年2期
牟 杨,杨志文
重庆大学附属涪陵医院 1.康复医学科;2.急诊医学部,重庆 408099
急性期重症脑损伤患者镇静镇痛药物选择尚未达成共识,68% ~76%存在不恰当的深镇静,导致神经损伤加重,谵妄发生率和病死率增加[1]。
右美托咪定(dexmedetomidine,DEX)可能是一个突破口,它兼具镇静、催眠、抗焦虑及镇痛疗效。已有动物实验表明DEX通过抑制兴奋性神经递质的释放,有明确的神经保护作用,但一直未能明确其细胞内机制,大大限制了DEX的临床推广应用。
轴突局部线粒体动力学(即线粒体分裂和融合)及线粒体轴突转运(mitochondrial axonal tran-sport, MAT)障碍是兴奋性神经递质过度释放的“触发点”。腺苷酸活化蛋白激酶(adenylate activates protein kinase,AMPK)是调节该“触发点”的关键信号蛋白,而DEX可激活AMPK信号通路[3],为确定DEX对受损细胞神经保护作用是否是通过激活AMPK实现的,观察了AMPK抑制剂compound C可否逆转DEX的神经保护作用。
因此,本研究通过建立肿瘤坏死因子-α(TNF-α)诱导的SH-SY5Y细胞模型,从线粒体角度探寻DEX产生神经保护作用的细胞内机制。
1 材料与方法
1.1 材料
人成神经细胞瘤细胞系SH-SY5Y(ATCC公司);抗KIF5B抗体(Cell Signaling公司);右美托咪定(DEX)、compound C(CC)、anti-OPA1、TNF-α(Sigma-Aldrich公司); 抗(anti-)p-AMPK、 anti-SNPH、anti-Drp1(Abcam公司);BCA 蛋白质测定试剂盒、细胞计数试剂盒、线粒体电位检测试剂盒、活性氧含量测定试剂盒(碧云天生物技术有限公司)。
1.2 方法
1.2.1 确定最佳TNF-α、DEX剂量:检测细胞系支原体污染,添加ATRA(10 nmol/L)分化6 d后,将细胞用于后续实验。于6孔板内接种细胞,待细胞培养至50%~70%汇合后,加10、20、50、100 ng/mL TNF-α,24 h后, CCK8比色法检测细胞存活率,将细胞存活率约为50%时TNF-α的剂量确定为后续实验中使用浓度。待TNF-α诱导的SH-SY5Y损伤细胞模型建立成功后,加10,20,50,100 nmol/L DEX,24 h后, CCK8比色法检测细胞存活率,将细胞存活率约为50%时 DEX的剂量确定为后续实验中使用浓度。
1.2.2 细胞的分组及处理:将细胞分为对照组(control)、TNF-α损伤细胞模型组(model,20 ng/mL,24 h)、DEX干预模型组(DEX,10 nmol/L,24 h)、DEX联合compound C干预模型组[DEX+CC(10 μmol/L)]4组。
1.2.3 Western blot检测细胞中p-AMPK、SNHP、KIF5B、Drp1、OPA1的表达:提细胞蛋白质并通过BCA法测定蛋白质浓度。用 12% SDS-PAGE 分离蛋白质,并转移到聚偏氟乙烯(polyvinylidene fluoride,PVDF)膜,5%脱脂奶粉室温封闭 2 h,分别加入p-AMPK、SNHP、KIF5B、Drp1、OPA1抗体于4 ℃过夜,室温下与人抗兔 IgG 孵育 1 h,化学发光试剂显影。用Image J 进行图像分析。
1.2.4 ELISA检测细胞中IL-1β和IL-6含量:根据试剂盒进行包被封闭、加样、孵育、显色,450 nm处测定光吸度值,在蛋白质含量标准化后,根据标准曲线计算炎性因子的含量。
1.2.5 用JC-1检测细胞线粒体膜电位(Δψm):将细胞接种在48孔板中,分别每孔滴加150 μL 10 μmol/L JC-1染料后,将48孔板放置于CO2细胞培养箱中,孵育30 min,后用JC-1染色;将48孔板放置在荧光显微镜下观察并留图。设定发射波长分别为590 nm、530 nm,测量并分析红、绿色荧光强度比。
1.2.6 用微孔板免疫捕获测定试剂盒检测细胞线粒体功能:按线粒体分离试剂盒说明书从细胞中分离线粒体。将线粒体匀浆加入相应的反应缓冲液中。转移到预热过的(30 ℃)石英比色皿中,complex Ⅰ、Ⅱ、Ⅲ、Ⅳ分别在450、600、550 和550 nm处测定吸光度,并在蛋白质含量标准化后,complex Ⅰ、Ⅱ、Ⅲ、Ⅳ活性以mA/min·mg protein表示。
1.2.7 用相应试剂盒检测细胞ATP、氧化指数:用比色法试剂盒检测总ATP水平(在570 nm处),并以μmol/mg蛋白为单位表示结果。按试剂盒测定细胞中超氧化物歧化酶(superoxide dismutase,SOD)活性、丙二醛(malondialdehyde,MDA)和谷胱甘肽(glutathione,GSH)含量。
1.2.8 用DCFH检测细胞内ROS水平:将细胞接种在48孔板,每孔滴加200 μL内含10 μmol/L DCFH和活细胞染色剂的混合液后, 放置于CO2细胞培养箱中,避光孵育20 min后洗涤;通过荧光显微镜观察绿色荧光强度,设定激发波长、发射波长分别为488 nm、525 nm。
1.3 统计学分析

2 结果
2.1 实验确定TNF-α和DEX最佳剂量
与对照组相比,模型组SH-SY5Y细胞存活率以剂量依赖性方式显著降低(P<0.001)(图1A),20 ng/mL处理24 h后,细胞存活率约 50%(P<0.001)(图1B),在后续实验中使用此剂量及时间点。随后,DEX(10~100 nmol/L)预处理 24 h后,与模型组相比,DEX 干预模型组(10~100 nmol/L)细胞存活率提高(P<0.001)(图1B),从患者获得DEX 血浆有效浓度为0.22~2.50 μg/L,对应浓度1.09~12.48 nmol/L,因此,后续实验中使用10 nmol/L DEX。

A,B.SH-SY5Y cells were treated with different concentrations of TNF-α and DEX. Cell viability was measured by MTT assay to determine the optimal dose; *P<0.001 compared with control group;# P<0.001 compared with model group;△ P<0.001 compared with DEX group.
2.2 DEX 减轻细胞线粒体功能损害、减少氧化应激及炎性反应
Model组较contol组complex Ⅰ~Ⅳ、ATP、GSH水平(P<0.001)(图2A~E,G), 和SOD (P<0.01)(图2H)水平显著降低,但MDA及IL-1β、IL-6水平显著增高(P<0.001)(图2F,I,J),DEX组较model组complex Ⅰ~Ⅳ及ATP、GSH、SOD水平显著增高,而MDA、IL-1β、IL-6水平显著降低(P<0.001)(图2A-J);DEX+CC组较DEX组complex Ⅰ~Ⅳ及ATP、GSH(P<0.01)(图2A~G)、SOD(P<0.001)(图2H)水平显著降低,而MDA、IL-1β、IL-6水平显著增高(P<0.01)(图2F,I~G)。
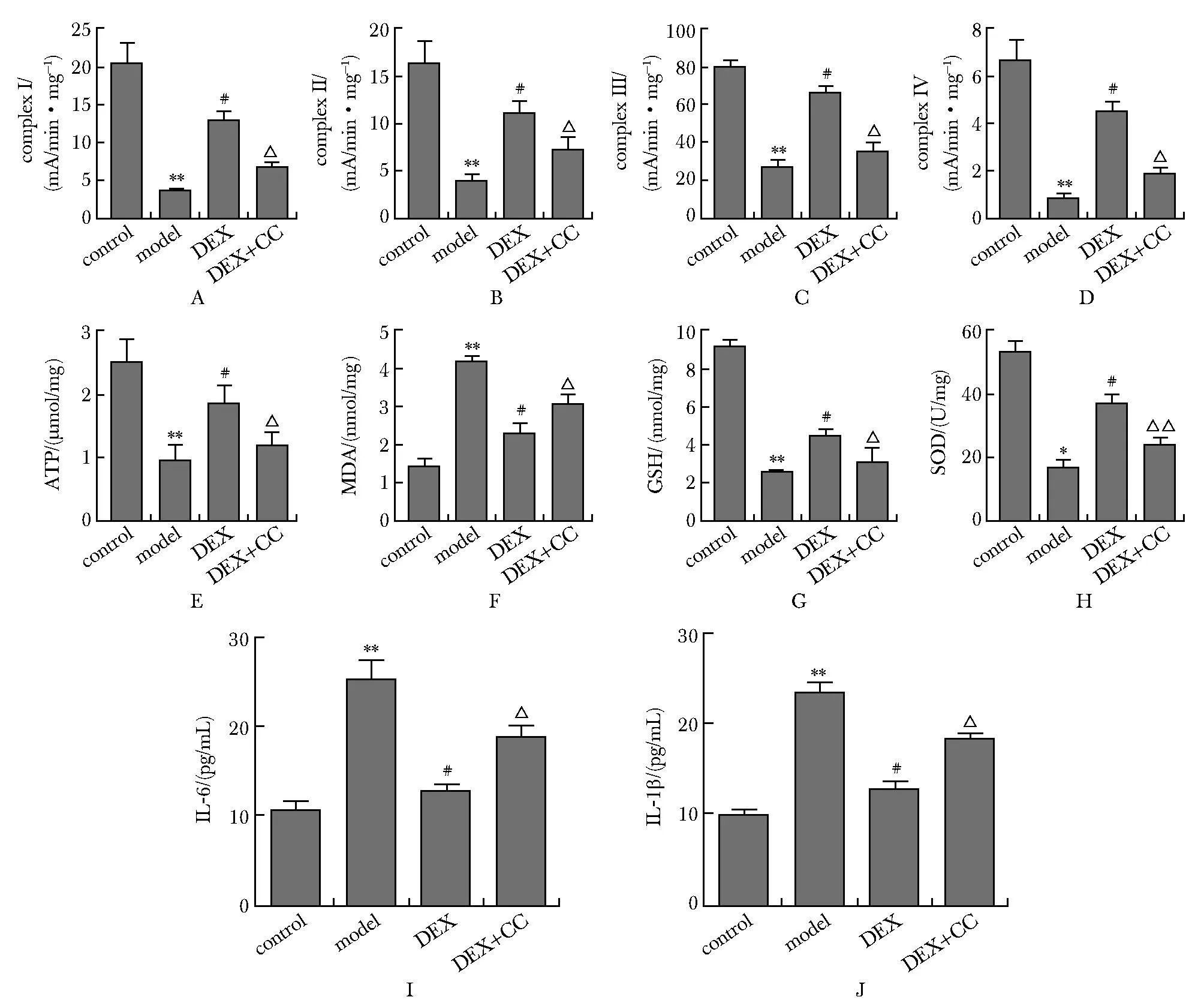
A-D.level of respiratory chain complex Ⅰ-Ⅳ detected by immunocapture assay; E.level of ATP;F.level of MDA;G.level of GSH;H.level of SOD; I.IL-6 content; J.IL-1β content; *P<0.01,**P<0.001 compared with control group;# P<0.001 compared with model group;△ P<0.01,△ △ P<0.001 compared with DEX group.
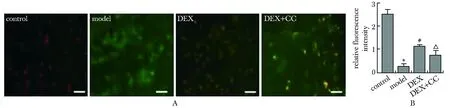
A.mitochondrial membrane potential(Δψm) was detected by JC-1 probe staining; B.quantitative analysis of Δψm; *P<0.001 compared with control group;# P<0.001 compared with model group;△ P<0.01 compared with DEX group.
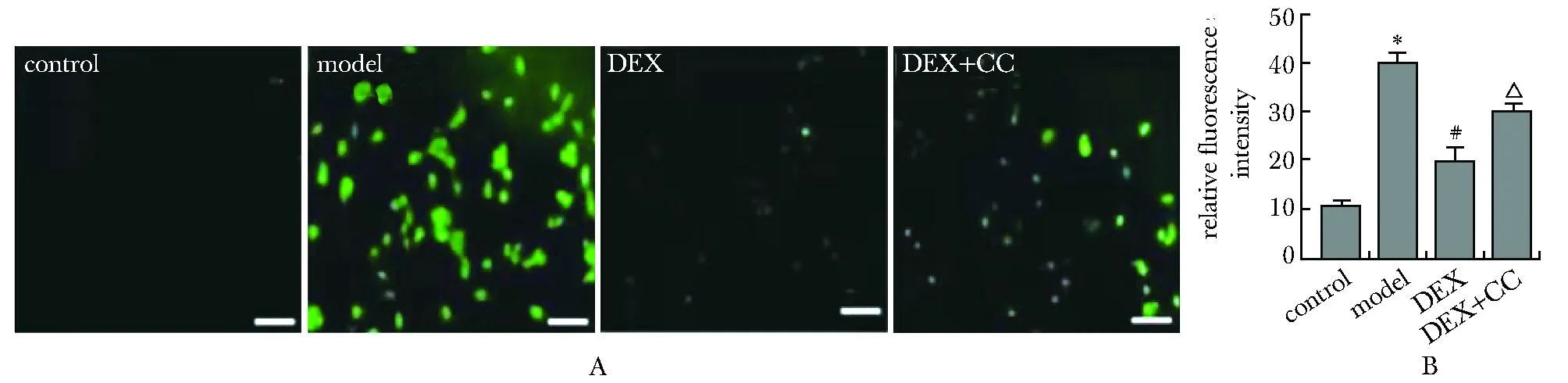
A.ROS assay detected by DCFH staining; B.quantitative analysis of DCFH staining; *P<0.001 compared with control group;# P<0.001 compared with model group;△ P<0.01 compared with DEX group.
2.3 DEX增加了模型组细胞中Δψm
Model组较contol组Δψm水平显著下降,DEX组较model组Δψm水平显著增高,DEX+CC组较DEX组Δψm水平显著降低(P<0.001)(图3A,B)。
2.4 DEX抑制了模型组细胞中ROS积累
Model组较contol组ROS水平显著增高,DEX组较model组ROS水平显著降低(P<0.001)(图4A,B),DEX+CC组较DEX组ROS水平显著增高(P<0.01)(图4A,B)。
2.5 DEX 可激活模型组细胞p-AMPK,上调OPA1和SNPH的表达,抑制Drp1和KIF5B的表达
Model组较contol组p-AMPK、SNPH及OPA1水平显著降低,而Drp1和KIF5B水平显著增高,DEX组较model组p-AMPK、SNPH及OPA1水平显著增高,而Drp1和KIF5B水平显著降低(P<0.001)(图5A~F),DEX+CC组较DEX组p-AMPK、SNPH(P<0.001)(图5A~F)及OPA1(P<0.01)(图5F)水平显著降低,而Drp1和KIF5B水平显著增高(P<0.001)(图5A~F)。

*P<0.001 compared with control group;# P<0.001 compared with model group;△ P<0.01,△ △ P<0.001 compared with DEX group.
3 讨论
TNF-α是重症颅脑损伤后导致神经炎性反应和线粒体功能障碍的重要细胞因子。本研究通过建立TNF-α诱导的SH-SY5Y细胞损伤模型,观察DEX对细胞损伤模型作用及其机制。发现DEX通过提高线粒体膜电位、呼吸链复合酶活性,促进ATP生成、抑制ROS产生等方面抑制TNF-α诱导的的线粒体功能降低,保护神经功能。
有研究表明,AMPK通过调节能量稳态和氧化应激,保护线粒体功能,抑制神经退行性变[4]。AMPK激活降低神经元介导的炎性因子水平[5],本研究发现DEX提高损伤模型细胞p-AMPK水平,与报道一致。同时发现AMPK激活是DEX产生抗伤害性保护作用的中心机制,AMPK抑制剂Compound C可逆转DEX对损伤细胞模型神经损伤的缓解,与报道一致[6]。
AMPK是线粒体动态平衡的关键调控因子[7]。而线粒体动力学(mitochondrial dynamics)及线粒体轴突运输(mitochondrial axonal transport, MAT)是[8]是正常细胞维持线粒体动态的关键[9]。因此,本研究假设激活AMPK可改善受损线粒体动力学,恢复局部能量供应,减少异常MAT,从而保护线粒体功能和抗氧化应激。
检测MAT主要调节蛋白——线粒体锚定蛋白SNPH和线粒体正向移动蛋白KIF5B的表达,发现DEX下调KIF5B表达,上调SNPH表达。有研究发现SNPH通过激活AMPK介导线粒体锚定作用,AMPK和SNPH的表达正相关[10]。SNPH通过与KIF5B结合介导线粒体活性依赖的固定,而AMPK磷酸化KIF5B并抑制线粒体向前运输。KIF5B表达减少导致线粒体正向转运减少,而SNPH的表达则会增加[11]。 在癌模型[12]中,SNPH降低导致氧化应激增加、细胞增殖降低和细胞线粒体运动增强。本研究假设和实验结果与上述报道一致:激活AMPK可通过下调KIF5B、上调SNPH来降低MAT,实现线粒体原位复能和抗氧化应激。
进一步检测线粒体动力学主要调节蛋白:裂变蛋白Drp1和融合蛋白OPA1的表达,发现DEX下调Drp1表达,上调OPA1表达,与报道一致[13]。
镇痛镇静在急性期重症脑损伤患者治疗中起着至关重要的作用,但目前临床上镇痛镇静药物多存在神经损伤作用。本研究显示,右美托嘧啶可能通过AMPK介导的抑制异常线粒体动力学,促进线粒体原位融合复能,对损伤神经细胞具有保护作用,可为急性期重症脑损伤患者镇痛镇静治疗提供更多思路。
猜你喜欢
核科学与工程(2022年3期)2022-10-18 01:25:14
建材发展导向(2021年11期)2021-07-28 06:57:22
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
广州化工(2020年6期)2020-04-18 03:30:20
当代水产(2020年10期)2020-03-17 07:02:48
当代水产(2019年8期)2019-10-12 08:57:26
现代矿业(2018年9期)2018-10-16 09:37:02
天然气与石油(2014年2期)2014-07-16 11:01:18
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
