
NK细胞代谢途径及其功能
2024-01-24 08:01于雅婷张建山东大学药学院免疫药物学研究所济南250012
中国免疫学杂志 2024年1期
于雅婷 张建 (山东大学药学院免疫药物学研究所,济南 250012)
NK细胞属于固有淋巴细胞(innate lymphoid cells,ILCs)成员,与其他ILCs起源于共同的淋巴样祖细胞。作为机体免疫防御的第一道防线,NK细胞可以在未经预先致敏的情况下快速杀死病毒感染的细胞和肿瘤细胞[1]。与B细胞和T细胞不同,NK细胞表面不表达重排的抗原特异性受体,而是由激活性受体和抑制性受体随机组合,两种受体信号动态平衡决定NK细胞对病原体的应答或耐受[2]。NK细胞的功能机制主要包括4方面:释放溶解颗粒穿孔素和颗粒酶产生细胞毒作用;分泌IFN-γ等细胞因子介导免疫调节和清除作用;上调死亡相关配体FasL、TRAIL等诱导靶细胞凋亡;通过抗体依赖的细胞介导的细胞毒性(antibody-dependent cellmedicated cytotoxicity,ADCC)效应杀伤靶细胞[3]。近年随着长寿NK细胞的发现,人们逐渐认识到NK细胞具有免疫记忆功能[4-5]。基于NK细胞快速的应答能力、独特的识别机制、强大的杀伤功能以及记忆特征,NK细胞已成为具有重要临床应用价值的免疫治疗细胞。
免疫系统在持续不断感知并清除病原体的过程中涉及巨大的能量消耗,这就需要免疫细胞从外部获得大量葡萄糖、氨基酸和脂肪酸等营养物质以维持供能。这些营养物质对免疫细胞的作用主要包括两方面:一是提供合成生物大分子RNA、DNA和蛋白质等的原材料以供细胞存活和增殖;二是为活化的免疫细胞提供合成ATP的底物以维持免疫应答[6]。因此,胞内代谢对免疫细胞稳定和发挥功能具有举足轻重的作用。不同代谢方式能够支持细胞多样的生物合成和功能发挥以维持机体稳态。当生物机体被病毒感染或肿瘤侵袭时,代谢往往会发生异常变化,造成免疫细胞功能失调,从而导致机体处于持续感染状态。研究发现,NK细胞的功能与其代谢息息相关,代谢重编程对NK细胞生长发育、记忆特征和免疫应答均发挥关键调控作用。因此,深入了解NK细胞代谢对促进NK细胞的抗肿瘤和抗病毒功能、开发和优化基于NK细胞的免疫疗法具有重要的理论意义和应用价值。
1 NK细胞的代谢方式
代谢是贯穿细胞生命周期的重要生理活动。在体内,糖、脂和氨基酸代谢以三羧酸(tricarboxylic acid,TCA)循环作为枢纽,构成复杂的代谢网络。细胞ATP的产生主要依赖于葡萄糖驱动的糖酵解和氧化磷酸化(oxidative phosphorylation,OXPHOS)。葡萄糖经糖酵解生成的丙酮酸进入线粒体后被氧化脱羧为乙酰辅酶A并进入TCA循环,产生还原当量NADH和FADH2,从而将电子传递到电子传递链(electron transport chain,ETC)复合物,使H+能够梯度穿过线粒体内膜,从而驱动ATP合酶活性[7]。此外,脂肪酸通过β-氧化产生的乙酰辅酶A及氨基酸分解产生的α-酮戊二酸(α-ketoglutaric acid,α-KG)也能够进入TCA循环,驱动ATP产生(图1)。
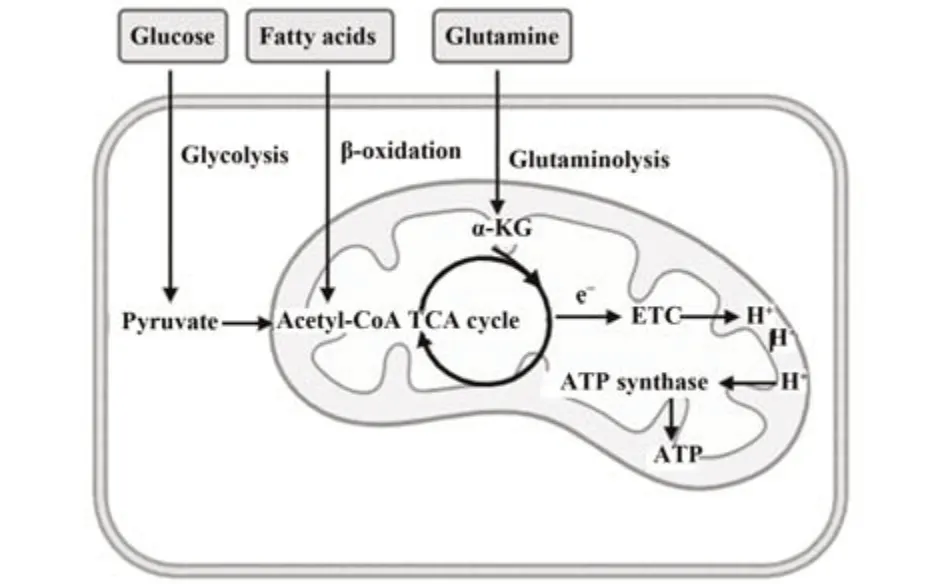
图1 NK细胞的代谢方式Fig.1 Metabolism of NK cells
1.1 NK细胞糖代谢 糖代谢是NK细胞最主要的代谢方式。静息状态下的NK细胞具有较低的基础代谢率,仅维持较低水平的糖酵解和OXPHOS速率。但持续刺激会导致NK细胞发生强大的糖代谢变化,为NK细胞发挥功能提供能量保障[8]。IL-2、1L-12和IL-18等细胞因子单独或联合刺激促进NK细胞对葡萄糖的摄取,提高糖酵解和OXPHOS速率[9];CD16抗体或NKG2D受体激活也可显著上调人NK细胞糖代谢速率,并促进脱颗粒分子CD107a和granzyme B表达[10]。通过糖代谢抑制剂可进一步说明糖代谢对NK细胞的重要性。糖酵解代谢抑制剂2-脱氧葡萄糖(2-deoxyglucose,2DG)和OXPHOS抑制剂寡霉素会明显抑制人和小鼠来源NK细胞代谢速率,并抑制IFN-γ分泌和颗粒酶表达[9,11];2DG还会降低IL-15激活的人NK细胞增殖能力和对靶细胞K562的杀伤能力[12];小鼠体内注射2DG会显著降低poly I:C诱导的NK细胞IFN-γ产生,并降低NK细胞对小鼠巨细胞病毒(murine cytomegalovirus,MCMV)的清除能力[12]。表明细胞因子或受体途径激活的NK细胞优先通过糖代谢途径提供发挥功能所需的能量。
1.2 NK细胞脂代谢 NK细胞是否使用脂质作为燃料还未进行深入研究。研究表明,脂肪酸氧化抑制剂对NK细胞IFN-γ和颗粒酶产生无明显影响,说明脂质可能不是NK细胞主要的能量来源[13]。但也有研究表明,未成熟的小鼠NK细胞表达更高水平的脂质合成相关酶,且记忆细胞形成需要脂肪酸氧化参与,说明脂代谢可能在NK细胞发育成熟初期及NK细胞记忆特征形成过程中发挥关键供能作用[14-15]。较为明确的是,体内脂肪酸过多堆积会阻碍糖代谢,并干扰NK细胞表型和功能[16]。
1.3 NK细胞氨基酸代谢 氨基酸不仅可以作为能源物质参与TCA循环,还可以维持代谢调节因子信号传导,因此维持细胞内氨基酸含量是NK细胞发挥功能所必需的。谷氨酰胺分解产生的α-KG可以为TCA循环提供燃料,促进ATP生成。谷胱甘肽是一种重要的抗氧化剂,对控制细胞氧化应激发挥核心作用。研究表明,体内谷胱甘肽缺乏能够增加脂质氧化损伤并抑制NK细胞糖代谢重编程,而外源性谷胱甘肽能够促进NK细胞效应[17-18]。此外,氨基酸转运蛋白SLC1A5和SLC7A5以及辅助亚基SLC3A2摄取谷氨酰胺和亮氨酸等也能够激活糖代谢通路,提高NK细胞糖酵解和OXPHOS速率[19]。
总之,葡萄糖驱动的糖酵解和OXPHOS是NK细胞主要的代谢途径,但糖、脂和氨基酸三大代谢方式在NK细胞生命活动中存在错综复杂的相互关系,脂代谢异常会造成NK细胞糖代谢失调和功能抑制,而氨基酸代谢对维持糖代谢稳定和灵活性发挥重要作用。
2 NK细胞代谢的关键调控因子
2.1 哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR) mTOR是一种非典型丝氨酸/苏氨酸蛋白激酶,是磷脂酰肌醇激酶相关激酶蛋白质家族成员。mTOR进化上相对保守,有mTORC1和mTORC2两种复合物存在形式[20]。研究表明,mTORC1在免疫细胞糖、脂和氨基酸代谢中发挥关键调控作用,是重要的代谢检查点。静息状态下的NK细胞具有较低的mTORC1活性,但IL-2、IL-12等细胞因子刺激可显著激活人和小鼠来源NK细胞中mTORC1信号通路,促进骨髓NK细胞增殖或外周NK细胞活化,上调糖酵解及OXPHOS速率以及IFN-γ和颗粒酶表达,而mTORC1抑制剂雷帕霉素则会显著削弱这些细胞因子对NK细胞代谢的促进作用[21]。此外,NKG2D配体刺激也通过上调mTORC1活性促进氨基酸转运蛋白SLC3A2表达和IFN-γ产生[9],证明mTORC1激活是NK细胞活化的先决条件。与mTORC1相比,目前针对mTORC2的研究较少。虽然mTORC2在NK细胞代谢调控中不发挥核心作用,但能够通过抑制STAT5通路降低氨基酸转运蛋白SLC7A5表达,削弱mTORC1活性,抑制NK细胞成熟和功能[22]。
2.2 c-MYC c-MYC是mTORC1的下游靶基因。作为一种重要的转录因子,c-MYC不仅可调控糖酵解相关酶表达,还能够上调NKG2D等激活性受体表达,促进NK细胞功能[23]。IL-2、IL-12或IL-15刺激后,激活的mTOR通过增加c-MYC表达上调NK细胞糖酵解和OXPHOS速率,c-MYC缺失显著减弱NK细胞对细胞因子的应答能力[24-25]。值得注意的是,细胞因子刺激NK细胞的最初阶段,c-MYC表达升高主要受mTORC1调控,但氨基酸的稳定性对其持续表达也发挥调节作用。研究表明,谷氨酰胺通过控制氨基酸转运体SLC7A5表达促进氨基酸摄取,氨基酸进一步通过促进c-MYC翻译或降解维持其在体内稳定表达。缺失谷氨酰胺或抑制SLC7A5会迅速降低NK细胞中c-MYC蛋白表达,导致糖酵解和OXPHOS速率降低[24]。此外,c-MYC作为内质网压力传感器IRE1α-XBP1的下游靶标,IRE1α-XBP1-MYC轴可调控NK细胞线粒体功能和OXPHOS速率,在NK细胞增殖和活化过程中发挥关键作用[26]。
2.3 甾醇调节元件结合蛋白(sterol regulatory element binding proteins,SREBP) 作为转录因子,SREBP也是mTORC1的下游靶基因,对NK细胞代谢和激活至关重要。SREBP能够诱导脂肪酸和胆固醇合成途径中诸多基因表达,包括脂肪酸合酶(fatty acid synthase,FASN)、低密度脂蛋白受体(low density lipoprotein receptor,LDLR)、乙酰辅酶A羧化酶(acetyl CoA carboxylase,ACC1)等,被认为是关键的脂质合成调节因子[27-28]。但SREBP调节的脂肪酸和胆固醇合成对NK细胞的活性并非主导作用。研究发现,细胞因子刺激NK细胞后,SREBP通过调节柠檬酸-苹果酸逆向转运蛋白SLC25A1和ACLY表达促进柠檬酸-苹果酸穿梭过程,使线粒体中产生的柠檬酸转运到细胞质,并被裂解成乙酰CoA和草酰乙酸,从而上调OXPHOS速率[29]。SREBP还能够通过调控c-MYC表达促进NK细胞中聚胺合成,提高糖酵解和OXPHOS速率,促进IFN-γ和颗粒酶表达,增强NK细胞对靶细胞的杀伤能力(图2)[30]。

图2 NK细胞代谢的关键调控因子Fig.2 Key regulatory factors of NK cell metabolism
3 NK细胞代谢与生理活动
3.1 NK细胞代谢与发育 NK细胞来源于骨髓,其发育需经历几个不同阶段。在小鼠中,共同淋巴样祖细胞产生共同淋巴样祖细胞前体,进一步发育为NK祖细胞。之后,未成熟的CD27+CD11b-NK细胞进一步分化为CD27+CD11b+NK细胞,并表达S1P5使其能够从骨髓向外周转移。外周成熟的CD27-CD11b+NK细胞通过其表面抑制性受体KLRG1表达而终止分化[3]。小鼠骨髓单细胞测序结果显示,CD27+CD11b-NK细胞的营养物质转运蛋白SLC3A2、SLC1A5和CD71,糖酵解相关酶HEXA、HEXB和PFKL以及脂质合成相关酶FASN、ACLY均呈高表达,以维持未成熟的NK细胞高水平增殖需求。随着不断发育,NK细胞增殖速度减慢,成熟阶段时CD27-CD11b+NK细胞达到静息状态,其转运蛋白和代谢相关酶表达随即降低[14]。与CD27-CD11b+NK细胞相比,CD27+CD11b-NK细胞具有更高的mTORC1活性,且缺失mTORC1基因的小鼠中成熟的NK细胞比例明显降低,进一步说明代谢参与小鼠NK细胞发育过程[22]。
人外周血NK细胞中主要存在两个亚群,CD56dimCD16+NK细胞具有更强的杀伤能力,而CD56brightCD16-NK细胞具有更强的分泌细胞因子潜能。目前普遍认为CD56brightNK细胞是更为成熟的CD56dimNK细胞前体细胞。研究表明,受到IL-2、IL-12或IL-15等细胞因子刺激时,CD56dimNK细胞葡萄糖转运体GLUT1、氨基酸转运体CD98表达以及mTORC1和c-MYC激活程度均明显强于CD56brightNK细胞,且CD56dimNK细胞具有更高的糖酵解和OXPHOS水平[9,31]。值得注意的是,NK细胞在发育过程中会经历驯化过程,其抑制性受体与配体结合从而实现功能成熟和自我耐受。研究表明,NK细胞在驯化过程中通过提高糖酵解和OXPHOS速率促进细胞增殖。与未驯化的NK细胞相比,经过驯化的人NK细胞中营养物质转运蛋白表达和糖酵解速率明显提高,而高代谢活性也满足了NK细胞发挥功能对能量的需求[32-34]。
虽然目前关于NK细胞发育各阶段的代谢特征尚无全面研究,但以上证据表明关键的代谢调节因子是NK细胞成熟的关键驱动因素,且成熟的NK细胞具有更高的代谢活性和效应功能。
3.2 NK细胞代谢与记忆 长期以来,免疫记忆一直被认为是适应性免疫独有的特征,但近年研究表明,NK细胞也会对病毒感染、半抗原和细胞因子刺激产生长期记忆反应[35]。此过程中,NK细胞会经历能量从密集到稳定的变化,其增殖减缓、功能减弱,从而导致静息的、长寿的且具有长期记忆的NK细胞产生。与初次激活的NK细胞相比,记忆样NK细胞经再刺激时能够利用其储备的能量产生更强大、更快速的反应。因此,胞内代谢在驱动记忆样NK细胞形成和应答中发挥重要作用。
研究发现,表达激活性受体Ly49H的小鼠NK细胞可以识别MCMV特异配体m157。MCMV感染初期(感染后7 d内),Ly49H+NK细胞能够快速扩增,导致线粒体膜电位(mitochondrial membrane potential,△φm)降低和线粒体ROS增加,并伴随线粒体自噬,以清除受损线粒体,保持线粒体稳定和适应性。感染后期(感染后8~28 d),Ly49H+NK细胞线粒体ROS连续下降,线粒体膜电位持续升高,并进入代谢平稳状态。之后,这些病毒特异性Ly49 H+NK细胞获得自我更新能力,可持续数月,再次激活时呈强大的抗病毒能力。使用mTOR抑制剂或AMPK激活剂诱导自噬能够通过调节线粒体代谢增加MCMV感染过程中记忆NK细胞数量[36]。研究表明,线粒体自噬诱导剂能够调节CD8+T细胞的脂肪酸氧化(fatty acid oxidation,FAO)过程,促进记忆细胞的发育,而自噬缺陷会导致线粒体FAO失调,抑制CD8+T记忆细胞的形成[37-38]。因此,线粒体自噬调节的OXPHOS和FAO在记忆样NK细胞形成过程中可能同样发挥关键作用。
有报道称,感染人巨细胞病毒(human cytomegalovirus,HCMV)的人外周血中持续存在一群共表达NKG2C和CD57的NK细胞亚群,这些细胞具有类似记忆特性,称为适应性NK细胞[39]。与非适应性NK细胞相比,适应性NK细胞mTORC1活性更强,线粒体呼吸能力和线粒体膜电位升高,糖酵解和OXPHOS速率也更高。来自血清HCMV阳性患者的适应性NK细胞对IL-12和IL-18等细胞因子刺激并不敏感,但对Fcγ受体依赖的抗体包被靶细胞的识别和反应能力更强[40]。进一步研究发现,适应性NK细胞的这种独特代谢特征和功能适应性受表观遗传学调控。适应性NK细胞中染色质修饰转录调控因子ARID5B表达上调,诱导编码电子传递链成分基因表达,增强线粒体代谢;同时,基因表达分析显示,HCMV特异的适应性NK细胞中脂质代谢相关基因表达也增加[15]。因此,燃料的多样化和灵活性可能有助于HCMV适应性NK细胞形成和功能,但多种代谢方式共同参与是否是HCMV适应性NK细胞形成的必要条件以及何种方式占主导地位仍需进一步探讨(图3)。
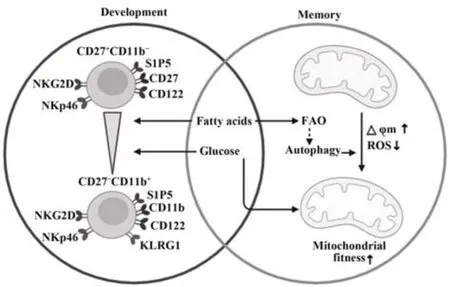
图3 代谢对NK细胞发育和记忆的调节机制Fig.3 Regulation of metabolism on NK cell development and memory
虽然上述研究初步证明了记忆性NK细胞形成和功能的代谢基础,但这些研究均受限于巨细胞病毒感染背景,其他病毒感染如乙型肝炎和流感病毒等也会诱导记忆样NK细胞产生[5,41],这些细胞的代谢特征及其与记忆功能形成的相关性仍有待确定。
3.3 NK细胞代谢与功能损伤 NK细胞功能与代谢息息相关。越来越多的证据表明,在癌症、慢性病毒感染和肥胖等一系列慢性疾病中,NK细胞代谢受损是导致NK细胞功能失调的一个重要原因。
3.3.1 肿瘤 NK细胞对肿瘤生长、侵袭和转移等发挥重要控制作用。然而,肿瘤细胞快速增殖会导致肿瘤微环境中葡萄糖、谷氨酰胺和氧气等营养物质缺乏,并积累大量腺苷、乳酸等代谢产物,造成NK细胞代谢障碍。研究表明,肿瘤微环境中NK细胞代谢改变可能是其功能受阻的一个重要因素。
3.3.1.1 缺氧 多数实体瘤快速增殖需要极大的耗氧量,导致肿瘤微环境中缺氧。缺氧会下调NK细胞NKp30、NKp46和NKG2D等激活性受体表达,并通过自噬途径降解颗粒酶,降低NK细胞效应功能[42-43]。研究发现,缺氧通过mTOR-Drp1途径诱导NK细胞线粒体过度分裂和碎片化,降低OXPHOS速率,损伤NK细胞的细胞毒活性,从而造成肿瘤免疫逃逸[44]。为应对缺氧环境、维持存活和功能,NK细胞通过缺氧诱导因子(hypoxia inducible factor,HIF)调节多种代谢基因表达。如短期缺氧环境诱导HIF-1α表达,IL-15和HIF-1α协同驱动NK细胞糖酵解基因表达,维持NK细胞功能[45]。但常氧情况下,HIF-1α基因缺失的NK细胞在IL-2和IL-12刺激下仍表现出正常的代谢和功能状态[24]。说明HIF-1α对NK细胞的调节与氧含量密切相关,对NK细胞在缺氧环境中的存活和功能至关重要。
3.3.1.2 代谢产物 即使在氧充足情况下,多数肿瘤细胞也会以糖酵解为主要供能方式,生长迅速的肿瘤细胞糖酵速率约比正常细胞高200倍,也就是所谓的“Warburg效应”。肿瘤细胞在大量摄取葡萄糖的同时产生过多乳酸,一方面阻碍了NK细胞对葡萄糖的利用,另一方面高浓度乳酸积累会造成极强的酸性肿瘤微环境,抑制NK细胞活力和功能。乳酸浓度大于20 mmol/L即可造成肿瘤内pH值偏低,导致NK细胞凋亡,可能是乳酸浓度较高的肿瘤组织中NK细胞数量较少的原因之一。结直肠癌肝转移的微环境中积累的乳酸能够引起肿瘤浸润肝脏内NK细胞pH值降低,导致线粒体应激,产生大量ROS,并诱导NK细胞凋亡[46]。同时,过多乳酸造成NK细胞过度酸化,抑制活化T细胞核因子(nuclear factor of activated T cells,NFAT)表达,使ATP合成受阻,导致NK细胞IFN-γ产生减少,抗肿瘤功能减弱[47]。乳酸也可通过调节NK细胞表面受体表达影响其功能。胰腺癌细胞中SIX1/LDHA途径诱导产生的乳酸会显著抑制NK细胞NKG2D、NKp46等活化性受体表达,并降低granzyme B、TNF-α水平[48]。在乳腺癌中通过阻断单羧酸转运蛋白-4(MCT4)减少乳酸外排、增加NK细胞pH值能够促进NKG2D表达,并提高NK细胞的杀伤能力[49]。
此外,肿瘤微环境中其他代谢产物,如吲哚胺2,3-二氧化酶(IDO)、腺苷、尿嘌呤等,也被证明能够抑制NK细胞功能。如乳腺癌细胞分泌的IDO能够抑制NK细胞ADCC效应[50];腺苷能够通过抑制NK细胞中IL-12和IL-15激活的STAT5和mTOR通路降低糖酵解和OXPHOS速率,损害NK细胞杀伤活性[51];尿嘌呤能够促进ROS产生,下调NKp46和NKG2D表达,引起人NK细胞凋亡[52]。一项最新研究表明,肿瘤细胞丝氨酸代谢失调会影响NK细胞表面鞘磷脂生物合成,抑制NK细胞膜突起形成,导致NK细胞无法形成免疫突触,进而失去杀伤肿瘤细胞的能力[53]。肿瘤微环境中代谢平衡对维持NK细胞活性至关重要。
3.3.1.3 TGF-β TGF-β是一种多效细胞因子,与多种肿瘤发生发展有关,并能够显著抑制NK细胞功能。研究表明,抑制细胞代谢是TGF-β诱导肿瘤微环境中NK细胞功能障碍的重要机制[54-55]。KRAS诱导的肺癌模型中,TGF-β诱导糖异生相关果糖-1,6-二磷酸酶(FBP1)在NK细胞中表达,FBP1通过抑制糖酵解损害细胞活力,引起NK细胞功能障碍,而抑制FBP1表达可以恢复NK细胞功能[56]。SLATTERY等[57]发现,转移性乳腺癌患者中,过多TGF-β会导致外周血NK细胞中mTORC1活性受损、线粒体形态改变以及OXPHOS速率降低,使NK细胞IFN-γ产生减少,死亡配体TRAIL表达降低,对肿瘤细胞的细胞毒活性减弱;而阻断TGF-β与其受体GARP结合可明显改善肿瘤微环境中NK细胞代谢活性及效应。
3.3.2 病毒感染 NK细胞长期暴露于慢性病毒感染的炎症环境中会呈现免疫无能状态。持续HIV感染会导致NK细胞线粒体去极化和碎片化,OXPHOS和糖酵解速率降低,使NK细胞对CD16抗体刺激的反应减弱,IFN-γ产生减少[58]。慢性乙肝患者外周血NK细胞mTORC1活性降低,EGR2、NR4A2和TOX等转录因子、LAG3和PD-L1等抑制性分子表达上调,呈现与耗竭T细胞相似的特征[59]。最近研究发现,抑制性受体表达增加与免疫细胞代谢受损有关。如PD-1能通过抑制氧化物酶体增殖物激活受体-γ共激活因子-1α(peroxisome proliferator activating receptor-gamma coactivator-1α,PGC-1α)表达,降低CD8+T细胞葡萄糖摄取、糖酵解速率和线粒体活性,驱动CD8+T细胞早期耗竭[60]。LAG3通过抑制AKT和STAT5通路阻碍CD4+T细胞糖酵解和OXPHOS速率,减少ATP产生,限制CD4+T细胞增殖[61]。由于在慢性病毒感染过程中,LAG3、PD-L1、Siglec-9和Tim-3等多种抑制性受体均在NK细胞表达上调,提示这些抑制性分子可能通过代谢途径阻碍NK细胞功能[62-63]。
3.3.3 肥胖 小鼠和人类研究均证明肥胖会增加癌症和感染发病率,在一定程度上与肥胖导致NK细胞功能障碍有关。饮食诱导或转基因肥胖模型小鼠感染流感病毒后,脾和肺组织中NK细胞数量和细胞毒性显著降低,更易因肺部感染而死亡[64]。肥胖儿童和成年人循环NK细胞比例降低,IFN-γ分泌较少,颗粒酶和穿孔素表达减少,且对肿瘤靶细胞的细胞毒性明显减弱[65]。MICHELET等[16]研究表明,肥胖通过诱导过氧化物酶体增殖物激活受体(PPAR)促进NK细胞大量摄取脂质,过多脂质积累抑制c-MYC和mTORC1信号诱导的代谢重编程,降低糖酵解和OXPHOS速率,导致NK细胞功能丧失。免疫突触形成是NK细胞精准杀伤靶细胞的重要条件,此过程需巨大能量消耗。与靶细胞形成免疫突触后,NK细胞线粒体膜电位迅速降低,达到代谢平稳状态,而肥胖小鼠及患有肥胖症人群NK细胞无法与靶细胞形成有效的免疫突触,可能与其线粒体代谢失调相关(图4)。
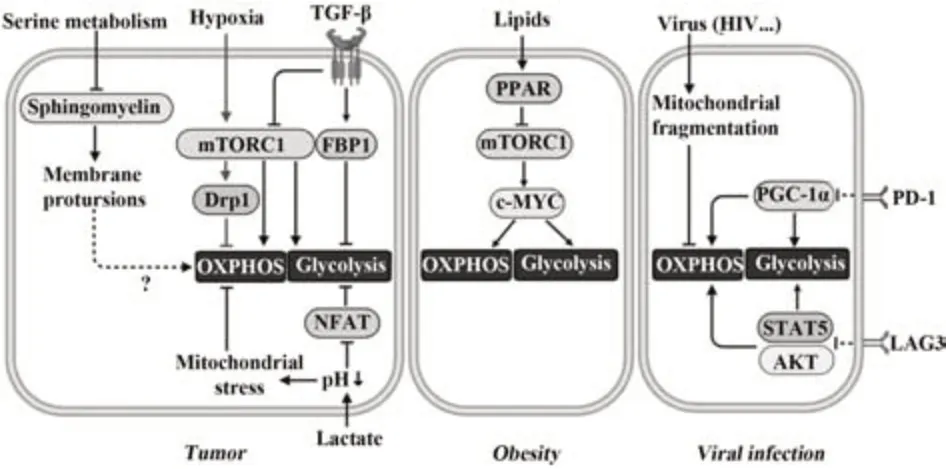
图4 慢性疾病中NK细胞代谢失调的调控机制Fig.4 Dysregulation of NK cell metabolism in chronic diseases
4 基于代谢的NK细胞疗法
了解活化NK细胞代谢需求有助于开发针对代谢的NK细胞免疫疗法。目前,能够通过改善代谢提高NK细胞功能的途径多样,包括通过使用小分子化合物、基因修饰、细胞因子疗法和干预免疫检查点等。
4.1 小分子化合物 糖原合成酶激酶3(glycogen synthase kinase 3,GSK-3)是一种高度保守的丝氨酸/苏氨酸蛋白激酶,可作用于多种底物,参与糖原代谢并调控细胞增殖与存活[66]。研究表明,GSK-3抑制剂能够通过抑制c-MYC降解促进NK细胞抗肿瘤应答[67-68]。尽管尚无证据表明GSK-3抑制剂的作用机制与代谢相关,但c-MYC通路在NK细胞代谢中的作用已被证实,提示GSK-3抑制剂可通过维持c-MYC活性改善NK细胞代谢,从而促进NK细胞抗肿瘤功能。ZHENG等[53]发现酸性和中性鞘磷脂酶抑制剂能够选择性阻断NK细胞中鞘磷脂分解代谢,促进膜突起和免疫突触形成,恢复NK细胞抗肿瘤活性。CB-839是一种靶向谷氨酰胺酶药物,目前正在晚期实体肿瘤患者中进行多个1期临床试验。CB-839能够减少肿瘤细胞对谷氨酰胺的消耗,使肿瘤微环境中谷氨酰胺含量增加,促进c-MYC信号传导,维持NK细胞糖酵解代谢[69-70]。同时,由于谷氨酰胺并非NK细胞的主要燃料,抑制其分解并不会阻碍NK细胞功能。另外,使用乳酸脱氢酶、吲哚胺2,3-双加氧酶1和环氧合酶2抑制剂等分别抑制肿瘤微环境中乳酸、尿氨酸和前列腺素等代谢废物产生,有利于提高肿瘤浸润NK细胞代谢灵活性[47,71-72]。除抑制剂外,针对一些特殊分子的激活剂对NK细胞代谢改善和功能调节也有重要作用。如RTA-408作为Nrf2抗氧化活性的有效激活剂,能够降低肿瘤浸润NK细胞氧化应激,提高NK细胞糖酵解和OXPHOS速率,促进其对肿瘤细胞的清除[17]。
4.2 细胞因子疗法 细胞因子对NK细胞代谢发挥关键调控作用。IL-2、1L-12、IL-15和IL-18等刺激均可上调NK细胞糖酵解和OXPHOS速率,其中IL-15对JAK/STAT和mTORC1通路具有强效激活作用,因此广泛用于改善NK细胞代谢适应性。不同肿瘤模型中,多种IL-15激动剂及重组IL-15蛋白单独或与其他治疗方法联用取得了显著抗肿瘤效果[73-74]。如小鼠重组IL-15与CD40抗体联用能够有效延长结肠癌小鼠生存期[75];IL-15与IL-15Rα受体连接的融合蛋白(SO-C101)能够显著减弱小鼠乳腺癌肺转移[76];重组人IL-15和IL-15激动剂N-803、H-803等均在临床抗肿瘤试验中取得了可观疗效[77-78]。此外,血液瘤特异CD123-CAR和CD19-CAR NK细胞中,同时过表达IL-15能够明显增强CAR-NK细胞活化和毒性,促进CAR-NK细胞对肿瘤的清除[79-80]。
4.3 基因修饰 TGF-β通过多种机制抑制NK细胞代谢,提示阻断NK细胞与TGF-β相互作用是一种有效提高NK细胞代谢速率的方法。最近研究证明,用显性失活的TGF-β受体进行基因修饰,NK细胞可抵抗TGF-β对NK细胞代谢的抑制作用,并增强其对胶质母细胞瘤和乳腺肿瘤细胞的杀伤能力[81]。细胞因子诱导的含SH2结构域的蛋白是细胞因子信号转导抑制因子家族成员之一,能够抑制NK细胞对细胞因子的应答,形成经典负反馈通路,防止细胞过度活化。ZHU等[82]发现,人诱导多能干细胞来源NK细胞中CIS基因敲除能够促进NK细胞对IL-15的响应,激活JAK/STAT和mTORC1通路,提高NK细胞糖酵解和OXPHOS速率,维持NK细胞存活和增殖,增强IFN-γ分泌、CD107a表达及对靶细胞K562的杀伤能力。此外,缺失免疫负调因子TIPE2(TNF-α-induced protein-8 like-2)、转录抑制因子ZHX2(zinc fingers and homeoboxes 2)等基因可促进小鼠NK细胞对IL-15的应答和mTORC1通路激活[83-84],其重要机制之一可能与改善NK细胞代谢适应性有关。
4.4 阻断免疫检查点 近年免疫检查点抑制剂已广泛用于逆转肿瘤或病毒感染导致的NK细胞功能障碍。多种肿瘤模型中,经典的抗PD-1或PD-L1抗体均可有效增强NK细胞抗肿瘤疗效[85-86]。近年研究发现,免疫检查点对免疫细胞代谢也有调节作用。LE等[87]发现PD-1通过刺激AMPK活性抑制糖酵解,并通过上调激活的CD4+T细胞犬棕榈酰基转移酶Ⅰ(CPT1A)表达促进脂肪酸氧化,从而抑制效应T细胞发育。PD-1或CTLA-4过表达会抑制T细胞受体刺激的葡萄糖和谷氨酰胺代谢上调[88]。此外,PD-1和PD-L1或PD-L2相互作用还会抑制PI3KAkt-mTORC1通路,破坏T细胞代谢重编程[89]。BENGSCH等[60]发现抗体阻断PD-1和PD-L1结合能够通过PGC-1α途径提高CD8+T细胞糖酵解和线粒体活性。因此,改善代谢可能是阻断免疫检查点逆转NK细胞功能障碍的机制之一。
5 结语
NK细胞作为机体监视和防御病原体的重要免疫细胞,深入研究其代谢与细胞功能活性的关系具有重要意义。虽然NK细胞代谢研究尚处于早期阶段,但目前研究指出代谢是贯穿NK细胞生命周期的重要活动,代谢重编程是NK细胞发育分化、记忆特征形成和发挥效应的必要驱动因素。未成熟的NK细胞通过高表达糖酵解相关酶和脂质合成酶,保持高水平代谢,维持快速增殖需求,驱动发育过程,发育成熟后细胞进入静息状态,代谢恢复到平稳水平。长寿需要极强的适应性和耐力,因此记忆NK细胞的形成通常伴随受损线粒体的清除、线粒体呼吸能力和膜电位的提高、损伤性活性氧水平的降低等过程,从而增强线粒体适应性。通过糖酵解和OXPHOS产生能量是NK细胞发挥功能的必要条件,而在肿瘤、肥胖和病毒感染等慢性疾病模型中,NK细胞由于糖代谢受限难以发挥有效的毒性能力。
尽管代谢对NK细胞调节已有大量研究成果,但仍有很多关键问题有待解答:①不同激活方式对NK细胞代谢的影响是否存在差异;②NK细胞通过不同机制发挥效应时其代谢需求是否一致;③血液循环NK细胞代谢特点是否适用于组织驻留NK细胞;④葡萄糖缺乏时,NK细胞优先使用何种燃料供能;⑤基于代谢的嵌合抗原受体等工程策略对NK细胞治疗是否有可行性。此外,由于不同功能的NK细胞缺乏特异的分子表达谱,难以精准区分,一些研究正在探讨根据代谢特征确定NK细胞亚群的可能性。但由于NK细胞燃料利用的灵活性、激活途径的广泛性、杀伤机制的多样性,以及NK细胞代谢调节涉及复杂的信号通路和分子,目前这些问题的解决还有极大的挑战性。值得期待的是,其他免疫细胞,如巨噬细胞和CD8+T细胞的代谢特征和调节机制可能对NK细胞具有参考性,将为针对NK细胞代谢进行更为详尽的研究提供有重要价值的依据。总之,NK细胞代谢是一个复杂且重要的研究领域,对NK细胞代谢的细致研究有助于深入了解其生理功能机制,并为开发基于NK细胞的免疫疗法提供新的指导策略。
猜你喜欢
中南医学科学杂志(2022年6期)2022-12-04
现代临床医学(2021年4期)2021-07-31
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中国循证心血管医学杂志(2020年11期)2020-01-08
中华老年口腔医学杂志(2016年4期)2017-01-15
中国医学装备(2016年6期)2016-12-01
中西医结合心脑血管病杂志(2016年20期)2016-03-01
医学研究杂志(2015年12期)2015-06-10
癌变·畸变·突变(2014年1期)2014-03-01
