
基于2-硫代巴比妥酸构筑零维原子簇{[Cu6(H2tba)6]·2DMF·xSolvent}、二维层状{[Sc(H2tba)3(DMF)]·2DMF}n和三维网状{[Fe(H2tba)2(H2O)2]·2DMF}n
2024-01-20 03:56:04何东美许嘉怡郑利民林悦健陈珍霞
无机化学学报 2024年1期
王 超 何东美 管 裕 许嘉怡 郑利民 林悦健 陈珍霞 梁 凯*,
(1东华大学化学与化工学院,上海 201620)
(2复旦大学化学系,上海 200433)
2-Thiobarbituric acid (H3tba)is a substituted mercaptopyrimidine with three mobile H atoms and has many possible tautomeric structures[1-2].H3tba and its derivatives are not only a kind of barbituric acid drugs,but also a polyfunctionalN,N′,O,O′,S-donating ligand with different binding centers, which can form novel complexes of diverse structures in the water phase[3-14].However,only a few reports have been devoted to the reaction of H3tba with metal ions in the organic phase[15-18].In this work, we provide three examples of using H3tba and three different metal ions for the assembly of a hexa-copper cluster complex {[Cu6(H2tba)6]·2DMF·xSolvent} (1), 2D sheet {[Sc(H2tba)3(DMF)] ·2DMF}n(2), and a 3D network structure coordination polymer {[Fe(H2tba)2(H2O)2]·2DMF}n(3).The luminescence properties of complex 1 were further studied.
1 Experimental
1.1 Materials and methods
All commercially available chemicals and solvents were of reagent grade and used as received without further purification.Sc(ClO4)3·6H2O was prepared by the reaction of Sc2O3and perchloric acid (70.0%-72.0%) in the fume cupboard.Caution: Perchlorates are potentially explosive and should be handled with great care and in small amounts.Elemental analyses for C, H, and N were carried out with a Vario EL Ⅲelemental analyzer.IR spectra were recorded on a Nicolet 6700 FT-IR spectrometer with KBr pellets in the 4 000-400 cm-1region.The thermogravimetric analysis(TGA)measurements were carried out on a TG 209 F1 thermal analyzer heated from room temperature to 900 ℃under N2atmosphere with a heating rate of 10 ℃·min-1.Powder X-ray diffraction (PXRD) patterns were taken on a Rigaku D/max-2550 PC X-ray diffractometer (CuKα,λ=0.154 184 nm) at room temperature(40 kV, 40 mA, 2θ=5°-70°).The fluorescent spectra were obtained on a PTI QM/TM luminescence spectrometer at room temperature.
1.2 Synthesis
1.2.1 Synthesis of complex 1
A solution of Cu(ClO4)2·6H2O (0.1 mmol, 37 mg)in MeOH (2 mL) was added to a solution of H3tba (0.2 mmol,29 mg)in DMF(6 mL),and colorless single crystals were obtained several days later.The single crystal diffracted weakly and easily lost some of the occluded solvents.The products were collected, washed with ethanol three times, and dried in air.Yield: 0.009 g(26% based on Cu).Anal.Calcd.for C24H18N12O12S6Cu6·4DMF·5CH3OH(% ): C, 29.09; H, 3.93; N, 13.24;Found(%): C, 29.35; H, 3.46; N, 13.63.IR (KBr, cm-1):3 104(s),2 973(m),2 976(m),1 654(s),1 637(s),1 526(s), 1 355 (s), 1 319 (s), 1 161 (s), 1 122 (m), 799 (m),520(m),458(m).
1.2.2 Synthesis of complex 2
A solution of Sc(ClO4)3·6H2O (0.25 mmol, 113 mg) in MeOH (2 mL) was added to a solution of H3tba(0.75 mmol,108 mg)in DMF(3 mL),and red rectangular plates formed.The products were collected, washed with ethanol three times, and dried in air.Yield: 0.058 g (52% based on Sc).Anal.Calcd.for C21H30N9O9S3Sc(%): C, 36.36; H, 4.36; N, 18.17; Found(%): C, 36.59;H, 4.83; N, 18.04.IR (KBr, cm-1): 3 146 (s), 3 071 (s),2 920(s),2 884(s),1 645(s),1 602(s),1 414(s),1 386(s),1 302(s),1 187(s),1 137(s),891(m),794(m),699(m),664(m).
1.2.3 Synthesis of complex 3
A solution of Fe(ClO4)3·9H2O (0.1 mmol, 52 mg)in MeOH (1 mL) was added to a solution of H3tba (0.2 mmol,29 mg) in DMF (0.5 mL),and yellow rectangular plates formed for single-crystal X-ray diffraction.The products were collected, washed with ethanol three times, and dried in air.Yield: 0.021 g (42% based on Fe).Anal.Calcd.for C14H24N6O8S2Fe(%): C, 32.07; H,4.61; N,16.03; Found(%): C, 31.94; H, 4.61; N,16.37.IR (KBr, cm-1): 3 108 (m), 3 078 (m), 2 874(m), 1 645(s), 1 597 (s), 1 201 (s), 1 182 (s), 890 (m), 815 (m),539(s),485(m).
1.3 Structure determination and refinement
Data collection for complexes 1 and 2 were carried out on a Bruker Smart Apex CCD diffractometer with a graphite-monochromatic MoKαradiation (λ=0.071 073 nm) at 173 and 273 K, respectively.Data collection for complex 3 was carried out on a Rigaku synergy X-ray single crystal diffractometer with CuKαradiation (λ=0.154 184 nm) at 100 K.The structures were solved by direct methods and refined onF2with the SHELX-97 program[19].All nonhydrogen atoms were refined with anisotropic thermal parameters.The positions of hydrogen atoms were generated geometrically.In complex 1, only two of the DMF molecules were refined isotropically with hydrogen atoms included.Due to the disorder, the other solvent molecules were removed using SQUEEZE[20].The structure of complex 3 is a merohedral twin.The crystallographic data and details of refinements for complexes 1-3 are summarized in Table 1.
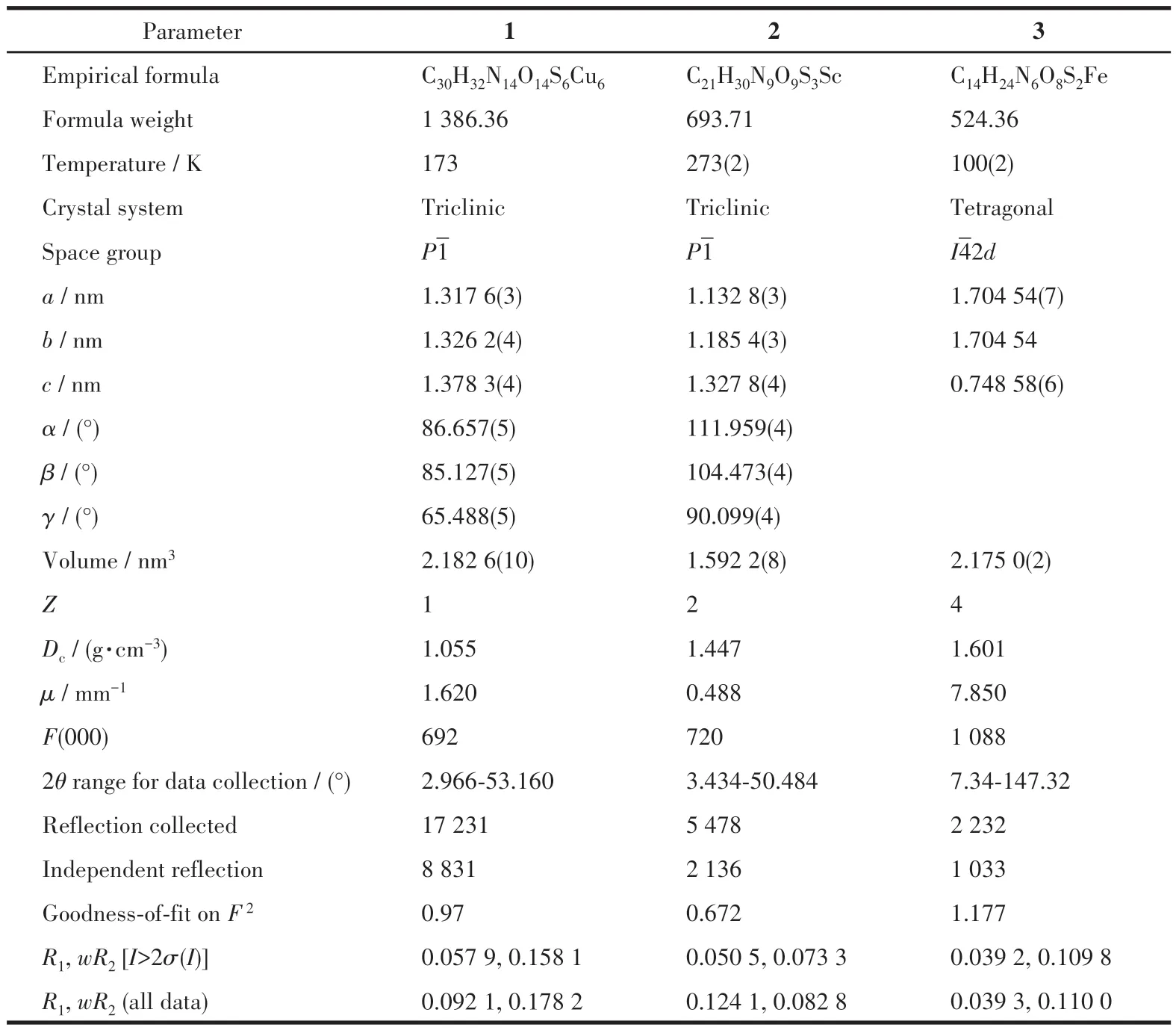
Table 1 Crystallographic data of complexes 1-3
CCDC:2298695,1;2298696,2;2298697,3.
2 Results and discussion
2.1 Crystal structure description
Single-crystal structure analysis of complex 1 reveals that the crystal structure is the triclinic system,space groupP1.The six copper atoms adopt a trigonal antiprismatic arrangement as shown in Fig.1a.Each copper atom is trigonally coordinated to a ligand nitrogen atom and two bridging sulfur atoms with bond lengths: Cu1—N5 0.200 5(4) nm, Cu2—N3 0.199 2(5)nm, Cu3—N1 0.199 8(4) nm, Cu1—S1 0.221 40(14)nm,Cu1—S3A 0.226 92(16)nm,Cu2—S2 0.223 27(14)nm, Cu2—S3 0.224 38(14) nm, Cu3—S1 0.221 52(15)nm, and Cu3—S2A 0.225 86(17) nm.Furthermore,each copper atom exhibits secondary Cu…Cu bonding to two other copper atoms forming a triangle(Cu1…Cu2 0.282 11(12) nm, Cu2…Cu3 0.292 19(12) nm, Cu3…Cu1 0.302 26(10)nm)with bond angles ∠Cu1—Cu2—Cu3 of 63.48(2)° , ∠Cu2—Cu1—Cu3 of 59.88(3)° ,∠Cu2—Cu3—Cu1 of 56.63(3)° indicated by broken lines in Fig.1a.In complex 1, the C—S bond lengths are in a range of 0.172 6(7)-0.175 3(6) nm, indicating they are single bonds.The ligand loses one proton, and the singly deprotonated ligand bridges the copper atoms through its sulfur and nitrogen donor atoms.Then Cu(Ⅱ)ions were found to result in a reduction to afford Cu (Ⅰ)ions in complex 1.The guest molecules desorb easily within a few minutes when the crystals are taken out from the mother liquor.There are large channels in the structure that contain disorder solvent molecules.The intermolecular hydrogen bonding and non-covalent interactions assemble these aggregates into a distorted face-centered cubic lattice, as illustrated in Fig.1b
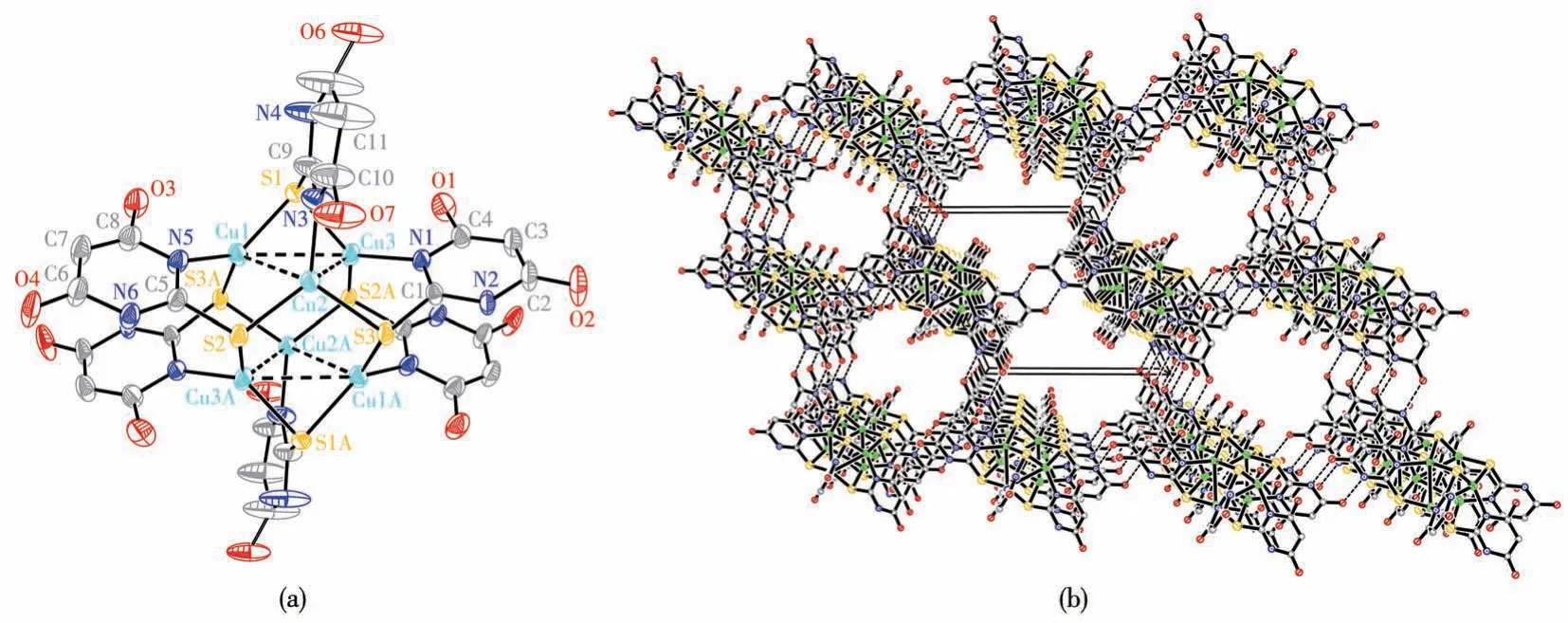
Fig.1 (a)Coordination environments of the Cu(Ⅰ)ions of complex 1 with thermal ellipsoid at 50% probability levels;(b)Packing diagram of 1 viewed along the a-axis
There are two different coordination modes for Sc(Ⅲ)ions in complex 2 as shown in Fig.2a.The Sc1 atom has six oxygen donors which consist of two O atoms from two DMF molecules, and four O atoms from four bridged H2tba-ligands.The other Sc2 atom has six oxygen donors which consist of two O atoms from two single H2tba-anions, and four O atoms from four bridged H2tba-ligands.The Sc1 atom is bound by six O atoms (O1, O1A, O4C, O4D, O5, and O5A),with bond lengths: Sc1—O1 0.211 1(3) nm, Sc1—O5 0.204 0(3) nm, Sc1—O4C 0.207 9(3) nm, and Sc1—O4D 0.207 9(3) nm, bond angles: O1—Sc1—O1A 180.0° , O4C—Sc1—O4D 180.0° , O5—Sc1—O5A 180.0° , O1—Sc1—O5 88.55(11)° , O1—Sc1—O4C 90.09(11)°, O5—Sc1—O4C 89.34(11)°, O5—Sc1—O4D 90.66(11)°.The Sc2 atom is bound by O2, O2B,O3, O3B, O7, and O7B atoms, with bond lengths:Sc2—O2 0.207 0(4) nm, Sc2—O3 0.204 2(3) nm,Sc2—O7 0.209 6(4) nm, bond angles: O2—Sc2—O2B 180.0° , O3—Sc2—O3B 180.0° , O7—Sc2—O7B 180.0° , O2—Sc2—O7 90.97(14)° , O2—Sc2—O3 91.22(13)°, O3—Sc2—O7 89.52(14)°.The Sc(Ⅲ)ions are bridged by four H2tba-anions to afford a 2D layer network viewed along the crystallographicc-axis, as illustrated in Fig.2b.The adjacent sheets are observed stacking with an ABC sequence.The 2D layer structure is stabilized by hydrogen bonds and aπ-πinteraction between the H2tba-ligands and DMF molecules.
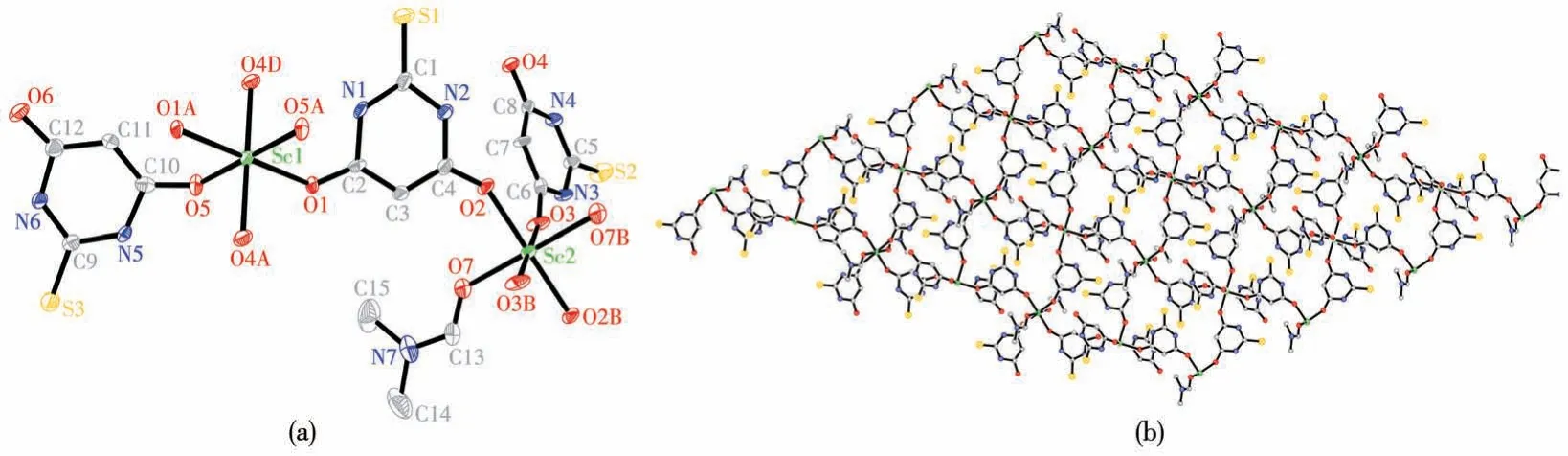
Fig.2 (a)Coordination environments of the Sc(Ⅲ)ions of complex 2 with thermal ellipsoid at 50% probability levels;(b)2D network of one sheet in 2 viewed along the c-axis
In complex 3, each Fe (Ⅱ) ion has six oxygen donors which consist of two O atoms from two coordinated H2O molecules, and four O atoms from four H2tba-ligands as shown in Fig.3a.The Fe (Ⅱ)ion is bound by O1,O1A,O1B,O1C,O1W,and O1WB atoms,with bond lengths:Fe1—O1 0.214 8(3)nm,Fe1—O1A 0.214 8(3) nm, Fe1—O1B 0.214 8(3) nm, Fe1—O1C 0.214 8(3) nm, Fe1—O1W 0.206 9(5) nm, and Fe1—O1WB 0.206 9(5) nm, bond angles: O1—Fe1—O1A 173.76(18)°, O1W—Fe1—O1WB 172.6(6)°, O1C—Fe1—O1B 173.76(18)°, O1—Fe1—O1B 90.170(11)°,O1—Fe1—O1C 90.170(10)° , O1W—Fe1—O1C 93.8(10)°, O1A—Fe1—O1C 90.170(11)°.In complex 3, the C3—S1 and C2—O1 distances are 0.166 2(5)and 0.127 1(5) nm, respectively, indicating they are double bonds.The C1—C2 distance is 0.139 5(5) nm,C3—N1 and the C2—N1 bond lengths are in a range of 0.135 7(4)-0.138 7(6) nm, indicating the C1 and N1 atoms are bonded with one H atom respectively.The H3tba ligand loses one proton, and the H2tba-ligand bridges the metal centers through two oxygen donor atoms.Then Fe(Ⅲ)ions were found to result in reduction to afford Fe(Ⅱ)ions in complex 3.There are large channels in the structure that contain the DMF molecules involved in the intermolecular hydrogen bonding that assembles these aggregates into a distorted facecentered cubic lattice,as illustrated in Fig.3b.

Fig.3 (a)Coordination environments of the Fe(Ⅱ)ions complex 3 with thermal ellipsoid at 30% probability levels;(b)Packing diagram of 3 viewed along the c-axis
2.2 TGA and PXRD
TGA performed for complex 1 as illustrated in Fig.4 showed a weight loss of about 27.63% at 265 ℃,which is close to the calculated values of 26.74% for the removal of guest DMF molecules and MeOH molecules.Complex 2 showed a weight loss of about 31.60% at 300 ℃, which is close to the calculated values of 28.15% for the removal of guest DMF molecules and coordinated DMF molecules.Complex 3 showed a weight loss of about 26.09% at 280 ℃, which is close to the calculated values of 24.70% for the removal of guest DMF molecules and coordinated H2O molecules.As the temperature increased, the elimination of the organic ligands began for complexes 1-3.The final residues (33.52%) for 1 at 900 ℃are almost agreed with the calculated values of 34.42% based on CuO.The final residues of about 25.50% for 2 at 900 ℃and 21.24% for 3 at 800 ℃are slightly higher than the calculated values of 19.88% and 13.70% based on Sc2O3and FeO, respectively.The reason could be attributed to the formation of Ln2O2SO4oxysulfates and oxide of iron.
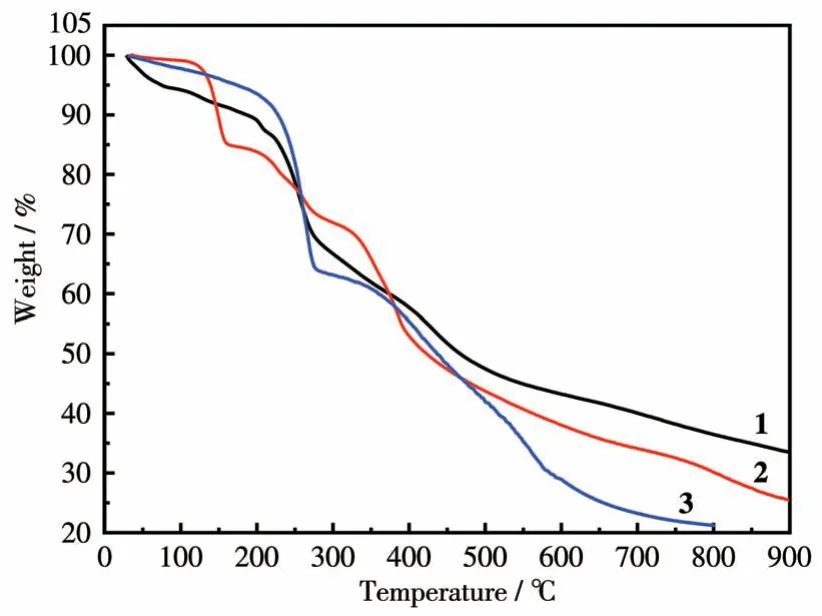
Fig.4 TG curves of complexes 1-3
The single crystals of complexes 1 and 3 lost crystallization when the crystals were taken out from the mother liquor a few minutes later.The loss of solvent molecules from the crystals may result in another phase.So, the PXRD analysis was performed for the complexes.The PXRD patterns of complexes 1 and 3 had slight displacement deviation from the singlecrystal simulation results.The single-crystal simulation result for complexes 2 agrees well with the experiment PXRD pattern,as illustrated in Fig.5.
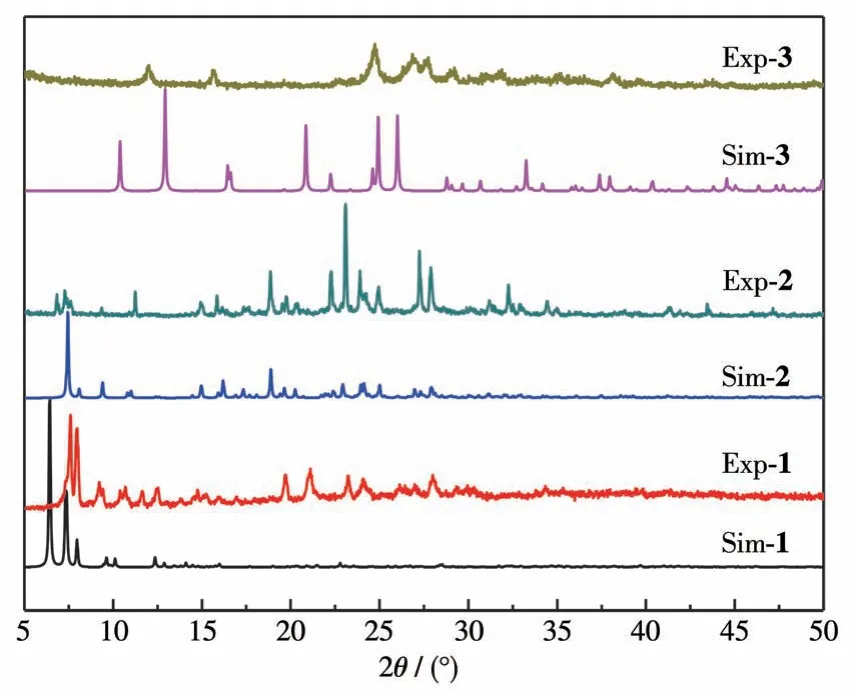
Fig.5 PXRD patterns of complexes 1-3
2.3 Luminescent property
The luminescent spectrum of complex 1 in the solid state is shown in Fig.6, with a maximum of 735 nm (λex=390 nm).Complex 1 exhibited an emission red shift compared with the free H3tba ligand upon the excitation, indicating that the emission of the complex may be assigned to the ligand-to-metal charge transfer(LMCT)[22].

Fig.6 Solid-state luminescence spectrum of complex 1 at room temperature
3 Conclusions
In summary, three complexes using tautomeric H3tba as ligands, with increasing dimensionality from the 0D cluster, 2D sheet, to the 3D network were synthesized by self-assembly reaction.The results reveal that the dimensionality of this kind of complex can be modulated by using different metal salts.We believe that diverse structure types can be further constructed with H3tba ligands in the future.
Acknowledgments:The authors gratefully acknowledged the financial support from the Fundamental Research Funds for the Central Universities(Grant No.2232014D3-10),and Shanghai College Students Innovation and Entrepreneurship Training Program.Prof.Kong Xiangjian at Xiamen University and Mr.Zhang Zaiyong at Shanghai Institute of Materia Medica, CAS were highly appreciated for their kind assistance in measuring single crystals.Prof.Ng Seik Weng at the University of Malaya was highly appreciated for his kind assistance in resolving the single crystal structure.
猜你喜欢
无机化学学报(2024年1期)2024-01-20 03:55:50
纺织服装周刊(2022年16期)2022-05-11 21:02:02
化工职业技术教育(2021年5期)2021-11-09 03:10:40
纺织科学研究(2021年1期)2021-03-19 05:18:14
化工职业技术教育(2021年1期)2021-03-15 06:57:44
纺织科学研究(2020年1期)2020-02-25 00:38:01
天然产物研究与开发(2018年5期)2018-06-13 03:23:46
中成药(2016年8期)2016-05-17 06:08:36
化工学报(2016年3期)2016-03-14 08:37:00
化工进展(2015年3期)2015-11-11 09:09:40
