
Cu2O-Au复合材料表面甲基橙降解过程的原位SERS研究
2024-01-17 03:40:42石灿姚建林
光散射学报 2023年4期
石灿,姚建林
(苏州大学材料与化学化工学院,苏州 215123)
1 引言
表面增强拉曼光谱(SERS)作为一种高灵敏、高化学特异性、高表面选择性的表界面研究技术,为贵金属催化剂催化反应的现场研究开辟了新途径[1]。众所周知,高质量的SERS光谱依赖于高质量的增强基底,早期主要以纳米级粗糙度的贵金属(如Au、Ag、Cu等)作为基底,在合适入射光激发下,基底表面局域电磁场得到巨大增强,表面吸附分子的拉曼信号得到显著增强[2]。但单一贵金属纳米粒子其实际功能性较为有限,一定程度上限制了SERS对表界面催化反应过程的研究,因此有必要将SERS技术拓展至复合型功能材料,将光学增强和催化等功能融合。
基于半导体材料的光催化是一种具有重要应用前景的绿色技术[3]。当自然光照射在半导体表面时,诱导电子-空穴对分离,光生电子、空穴与溶解在体系中的氧和水等物种反应生成活性更高的中间体,由此氧化分解水体中的有机污染物以达到净化水体的目的。不同类型的半导体金属氧化物如WO3[4]、ZnO[5]、Cu2O[6]等,非氧化物如CdS[7]、CuInS2[8]等,非金属半导体如C3N4[9]已被广泛用作光催化剂。其中,Cu2O作为一种窄带隙的p型半导体,其吸收光谱在可见光范围内,在光催化应用方面具有巨大的潜力。但单一的半导体材料往往存在着量子效率低、太阳能利用率差等问题,且其SERS效应较弱,而将贵金属沉积于半导体表面是解决这些问题的有效途径之一[10]。一方面,建立在界面上的肖特基结阻止了载流子通过内部电场和肖特基势垒进行复合[11];另一方面,贵金属的LSPR效应增加了对太阳能的吸收,为载流子运输提供了快速通道[12]。
在原位监测反应过程中,既可以利用基底的光催化活性诱导催化反应,又可以通过基底的SERS活性实现从分子水平上解析反应物以及产物结构,为催化过程研究提供更为精准的分子指纹信息。本文通过湿化学法制备了Cu2O-Au复合材料,以甲基橙(MO)光催化降解为模型反应,开展了对MO光催化降解性能以及过程的在线监测,深入研究了光催化的分子机制。
2 实验部分
2.1HAuCl4·4H2O、NaBH4、抗坏血酸、CuCl2·2H2O、NaOH、聚乙烯吡咯烷酮(PVP,Mw=10000;k30)、无水乙醇、均为分析纯,购自国药集团;甲基橙购自源叶生物,纯度为分析纯。由日本日立公司的S-4700型冷场发射扫描电镜测试SEM以及EDS谱;TEM图像采自日本日立公司的HT7700透射电子显微镜。XRD图谱在德国布鲁克生产的D8 Advance型X射线衍射仪下测试得到,测试的2θ范围为20-80°;采用Horiba Jobin Yvon生产的XploRA PLUS型拉曼光谱仪测试拉曼光谱,激发波长为638 nm,采用50倍长焦物镜或100x物镜,光栅为1200 g/mm。
2.2 Cu2O-Au复合材料的制备与表征
八面体Cu2O及Cu2O-Au的制备[13]:将1.5 g Mw=10000的PVP添加到100 mL 0.1 mol·dm-3的CuCl2·2H2O水溶液中,匀速滴入10 mL 2 mol·dm-3的NaOH水溶液,搅拌30 min后,溶液变为深棕色,匀速滴入10 mL 0.6 mol·dm-3抗坏血酸水溶液,将混合物陈化3 h,溶液逐渐由深棕色转化为砖红色,上述所有过程均在55 ℃水浴中进行,通过离心收集沉淀物,用蒸馏水和无水乙醇分别洗涤三次,置于50 ℃真空烘箱干燥6 h备用。将3.5 mg上述制得的八面体Cu2O与0.5 g PVP(Mw=10000)溶于10 mL乙醇中,超声5 min使其混合均匀,随后滴加2.5 mL 0.6 mol·dm-3HAuCl4水溶液,搅拌10 min后加入2.5 mL 3 mmol·dm-3NaBH4水溶液,搅拌30 min后离心收集沉淀物,采用蒸馏水和无水乙醇分别洗涤三次,置于50℃真空烘箱干燥6 h即可得到八面体Cu2O-Au。
3 结果与讨论
3.1 Cu2O及Cu2O-Au的形貌与结构
所制备的八面体Cu2O及Cu2O-Au的形貌如图1所示,由图可见八面体Cu2O形貌规整,大小均一,尺寸分布在1 μm左右,边缘较为光滑(图1a, b, c)。当其表面覆盖Au壳层后,Cu2O的表面变得粗糙,覆盖了一层粒径极小的Au纳米粒子层,其尺寸在5 nm左右,这是强还原剂NaBH4参与反应所致(图1d, e, f)。
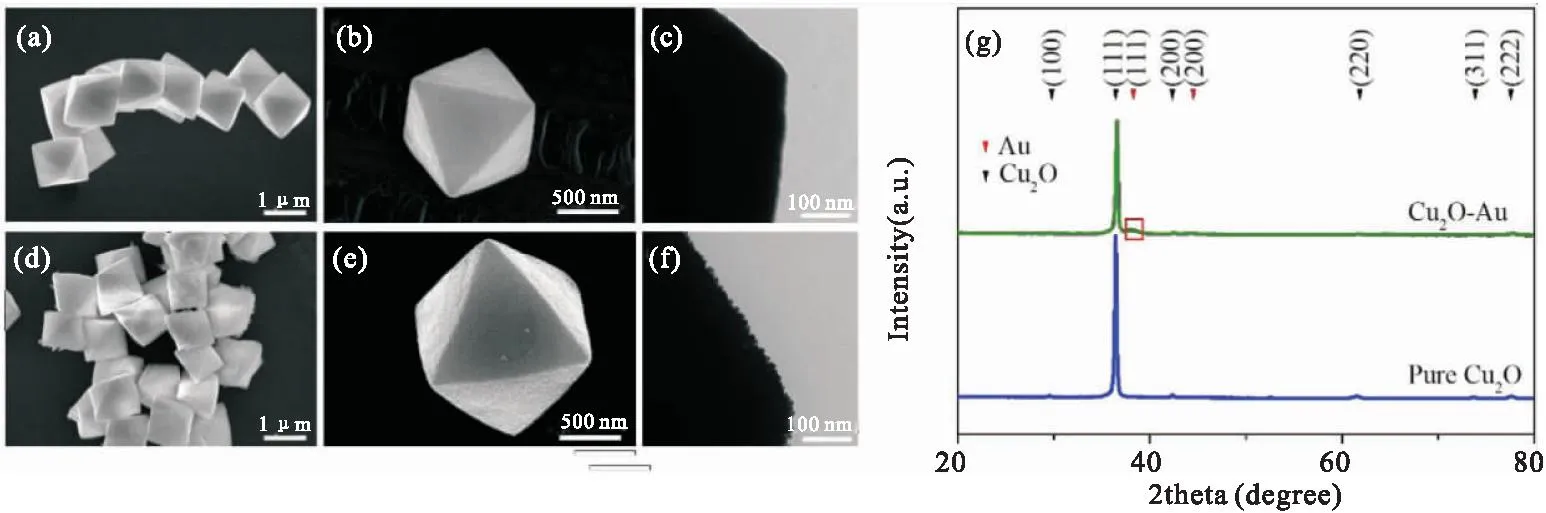
图1 八面体Cu2O的SEM(a、b)以及TEM(c)图像;八面体Cu2O-Au的SEM(d、e)以及TEM(f)图像;八面体Cu2O及Cu2O-Au的XRD图谱(g)Fig.1 The SEM images of octahedral Cu2O (a、b) and octahedral Cu2O-Au (d、e) , the TEM image of octahedral Cu2O (c) and octahedral Cu2O-Au (f) and their XRD spectra (g)
图1(g)为Cu2O和Cu2O-Au的XRD光谱。由Cu2O的X射线标准卡片(JCPDS No.05-0667)可知,在2θ为29.5°、36.4°、42.3°、61.4°、73.5°和77.4°位置上出现了六个主要特征X-射线衍射峰,分别对应(110)、(111)、(200)、(220)、(311)和(222)六个晶面衍射峰,这与图中衍射峰出现的位置一致,说明制得的Cu2O材料纯度高。由Au的X射线标准卡片(JCPDS No.04-0784)可知,Au峰位于38.2°和44.4°,分别对应于其(111)和(200)晶面。由以上XRD图谱可知,Cu2O-Au的XRD谱都同时包含了Cu2O和Au的特征衍射峰,但Au的特征衍射峰峰强较小,结合TEM图,这表明了Au纳米粒子成功地包覆在Cu2O上,但含量较低。
3.2 Cu2O-Au复合材料的SERS活性
图2为TP分子在Cu2O和Cu2O-Au基底表面的SERS光谱,所用激光功率0.13 mW,积分时间为20 s,采用100x物镜。由于在显微镜下可清晰观察到八面体Cu2O和Cu2O-Au,因此所有研究均仅针对单个Cu2O和Cu2O-Au粒子。如图2a所示,在Cu2O、Cu2O-Au表面均清晰地检测到位于1000 cm-1、1023 cm-1处归属于TP分子的环呼吸振动峰以及1073 cm-1、1575 cm-1处分别归属于TP分子的υ(C-S)谱峰和苯环上的υ(C=C)谱峰,且其SERS谱峰的位置和相对强度并未出现明显差别,但其谱峰绝对强度发生了显著变化,Cu2O-Au表面较Cu2O表面TP的SERS谱峰强度增强了约两个数量级,同时也说明了将SERS增强区域和催化反应活性区域完全统一,SERS信号真实反应了复合物的催化行为。
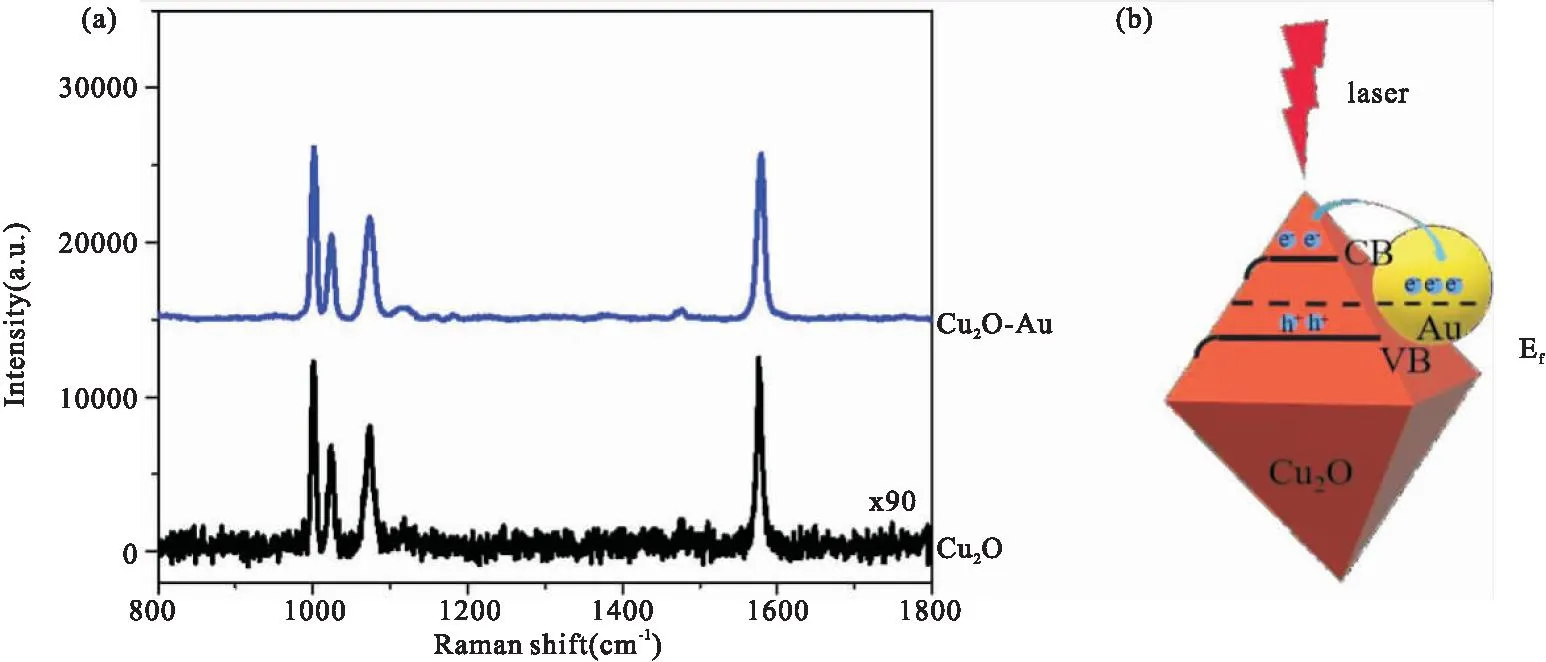
图2 八面体Cu2O、Cu2O-Au基底表面TP的SERS谱图(a)及激光照射下Cu2O-Au中的电子转移示意图(b)Fig.2 SERS spectra of TP adsorbed on octahedral Cu2O and Cu2O-Au (a) ; Schematic diagram of electron transfer under laser illumination in the Cu2O-Au (b)
SERS增强主要来自电磁场增强和化学增强,其中前者占主导作用。对于半导体Cu2O来说,其SERS效应的主要贡献来自于化学增强,增强效应相对较弱。对于Cu2O-Au复合材料,一方面,Au纳米粒子的覆盖使复合材料在入射光激发下可产生具有长程效应的SPR,SPR产生的表面局域电磁场显著地增强了吸附分子的SERS信号[14];值得说明的是表面覆盖的Au纳米粒子尺寸较小,对SERS效应并非十分有效,但有利于充分发挥Cu2O和Au界面处的性能,并提供足于检测到界面处反应的SERS增强效应;另一方面,如图2(b)所示,Au纳米粒子和Cu2O紧密接触形成肖特基势垒,激光照射下,自由电子自发从Cu2O流向Au纳米粒子,且Au纳米粒子表面的SPR发生退激产生热电子,肖特基势垒的存在阻止金属端的热电子重新流向半导体端,热电子在Au纳米粒子表面累积,使得复合材料上的吸附分子的SERS信号增强[15]。
3.3 Cu2O-Au复合材料的光催化活性
取6 mg的Cu2O和Cu2O-Au分别加入30 mL 0.1 mmol·dm-3的甲基橙(MO)水溶液中,在超声的辅助下,自然光照射每隔15 min取3 mL上述两种溶液进行离心,取上层清液UV-Vis测试,总降解时间为120 min(如图3所示)。
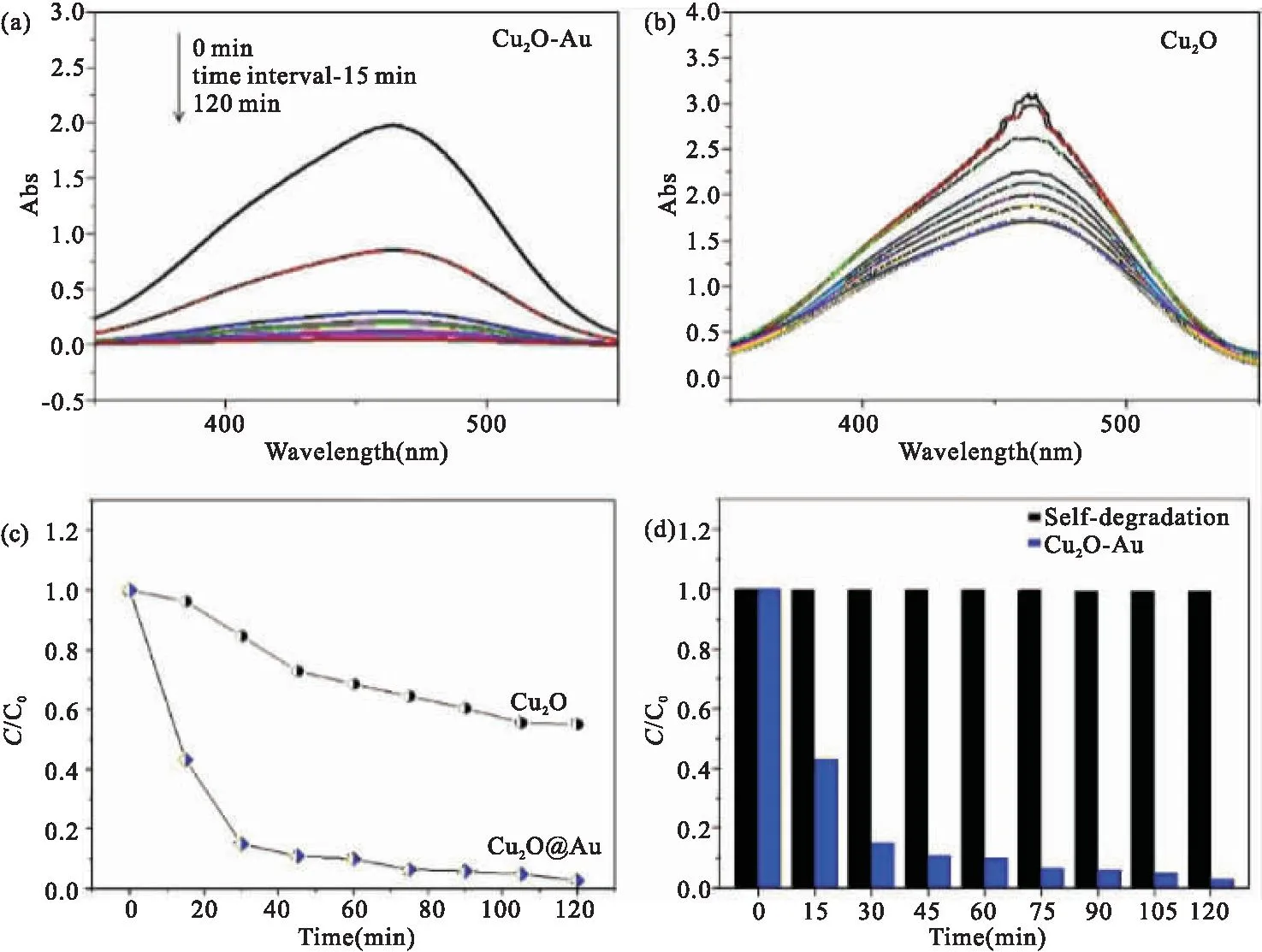
图3 Cu2O-Au(a)、Cu2O(b)作催化剂时MO溶液浓度随时间变化的UV-Vis光谱及其对应随时间变化的MO溶液浓度变化(c);自然光照下MO自降解以及Cu2O-Au催化降解随时间变化的MO浓度变化(d)Fig.3 Time-dependent UV-Vis spectra of MO catalyzed by Cu2O-Au (a) and Cu2O (b) ; and the corresponding MO solution concentration - time profiles (c) ; Time-dependent self-degradation of MO and degradation of MO catalyzed by Cu2O-Au under natural light (d)
图3a和b分别为Cu2O-Au复合材料和单一Cu2O在自然光状态下光催化降解MO的UV-Vis光谱。如图3a所示,MO位于465 nm处的最大吸收峰随自然光照射时间延长其强度逐渐降低,这说明自然光照下,Cu2O-Au可有效催化降解MO。在Cu2O体系中,随着自然光照射时间的延长,MO的紫外吸收峰变化并不明显(如图3b所示),这说明在单一Cu2O作为催化剂时,仅有部分MO被降解,图3c为分别以单一Cu2O和Cu2O-Au作为催化剂光催化降解MO时,降解前后MO浓度之比随时间的变化趋势。当光照射至120 min时,Cu2O-Au催化MO的降解率达到了97%,而单一Cu2O作催化剂,降解率仍然不到45%,单一的Au纳米粒子催化降解率仅约8%。图3d为MO在自然光状态下自降解和Cu2O-Au作催化降解120 min前后浓度之比,该时间内MO的自然降解程度不到1%。由此可见,Cu2O-Au复合材料展现出了优异的MO光降解性能,约为Cu2O的2倍,主要源于Cu2O-Au形成的界面。
Cu2O作为一种p型半导体具有较窄带隙(Eg=2.17 eV),对可见光有着良好响应性能,但单一的Cu2O催化剂的载流子分离效率极低,严重阻碍了它的实际应用,通过将Au纳米粒子沉积在Cu2O得到Cu2O-Au复合材料,一方面,其表面的Au纳米粒子提高了材料的比表面以及对光的吸收能力[16];另一方面,Au纳米粒子和半导体Cu2O之间形成异质结,异质结的存在阻碍了电子空穴对的复合,解决了单一Cu2O材料载流子分离效率低的问题,使体系中热电子、热空穴的数量有效增加,产生更多活性物质驱动降解反应进行[17]。
3.4 SERS在线监测MO降解
图4为甲基橙粉末的拉曼谱峰和吸附在Cu2O-Au基底表面的SERS谱峰,根据文献对甲基橙粉末的常规拉曼谱峰进行指认[18],如表1所示。在800 cm-1到1800 cm-1区域,位于1120 cm-1、1141 cm-1、1194 cm-1处的峰分别归属于δ(C-C)、υ(Ph1-N=)、υ(=N-Ph2)谱峰,位于1311 cm-1、1361 cm-1、1389 cm-1处的峰分别归属于δ(C-N)、υ(Ph-N)、υ(N=N)和υ((C-)SO2(-O))的合谱,位于1410 cm-1、1418 cm-1、1441 cm-1处的峰分别归属于δ(C-H)、υ(C-C)、υ(N=N)谱峰,位于1588 cm-1的峰归属于苯环上的υ(C=C)谱峰。根据其常规拉曼谱峰对MO的SERS谱峰进行归属,可发现二者光谱特征差异并不十分显著,仅有拉曼位移的细微变化,这些差异是因MO在基底上的吸附差异所致。
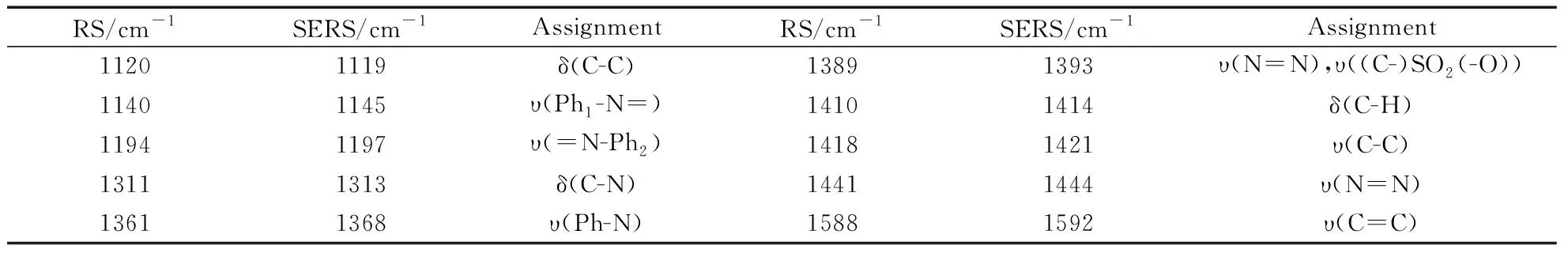
表1 MO的常规拉曼光谱峰以及SERS光谱峰归属Table 1 The normal Raman and SERS spectrum peak assignment of MO
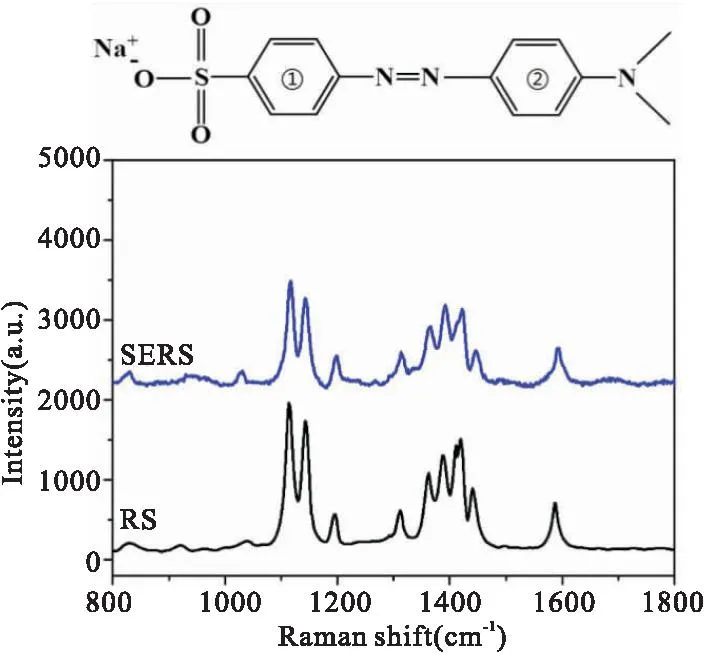
图4 甲基橙粉末的常规拉曼光谱及其在Cu2O-Au基底表面的SERS光谱Fig.4 The normal Raman spectrum of MO and its SERS spectrum adsorbed on Cu2O-Au surface
取6 mg Cu2O-Au加入30 mL 0.1 mmoldm-3的MO水溶液中,在自然光照射下每隔20 min取3 mL溶液离心后进行拉曼测试,激光功率为13.8 mW,积分时间为10 s。如图5a所示,在降解至60 min时,位于1441 cm-1处归属于MO的υ(N=N)谱峰消失,位于1194 cm-1处归属于MO的υ(=N-Ph2)谱峰发生蓝移;当降解进行至120 min时,上清液的拉曼谱峰呈现出与空白基底背景一致的谱峰特征,这说明此时MO几乎被完全降解。以甲醇为流动相,流速为1 mL·min-1,使用HPLC对上述上层清液进行分离,每组样品进样量均为10 μL。图5b为0.1 mmoldm-3MO水溶液以及Cu2O-Au光催化降解MO第60 min、120 min的色谱图。甲基橙标准溶液在色谱峰位于1.7 min左右,整个降解过程120 min,MO色谱峰面积降低了92.5%,但整个降解过程中并未检测到有中间产物。

图5 Cu2O-Au作催化剂时MO随时间变化的拉曼谱图(a);0.1 mmoldm-3 MO水溶液以及Cu2O-Au光催化降解MO第60 min、120 min的分离色谱图(b)Fig.5 Time-dependent normal Raman spectra of MO catalyzed by Cu2O-Au (a) ;Chromatograms of 0.1 mmoldm-3 MO aqueous solution and Cu2O-Au photocatalytic degradation of MO for 60 min and 120 min (b)
图6为MO位于800 cm-1到1800 cm-1的波数区域内随时间变化的SERS谱峰,随着激光的照射,除位于1592 cm-1处归属于MO苯环上υ(C=C)谱峰峰强度未发生明显变化外,其余MO的各峰峰强逐渐下降,这说明,苯环上C=C难以断裂。激光照射时间至3 min时,观察到了位于1237 cm-1处归属于对位二取代苯的环振动谱峰,且随着激光照射时间的延长,该峰逐渐增强。对比降解10min前后的SERS谱图,可明显发现,位于1414 cm-1、1421 cm-1处分别归属于MO δ(C-H)、υ(C-C)谱峰强度下降速度明显要快于位于1368 cm-1处归属于MO的υ(Ph-N),这说明C-H、C-C对催化剂Cu2O-Au更为敏感,相较于Ph-N更容易断裂。
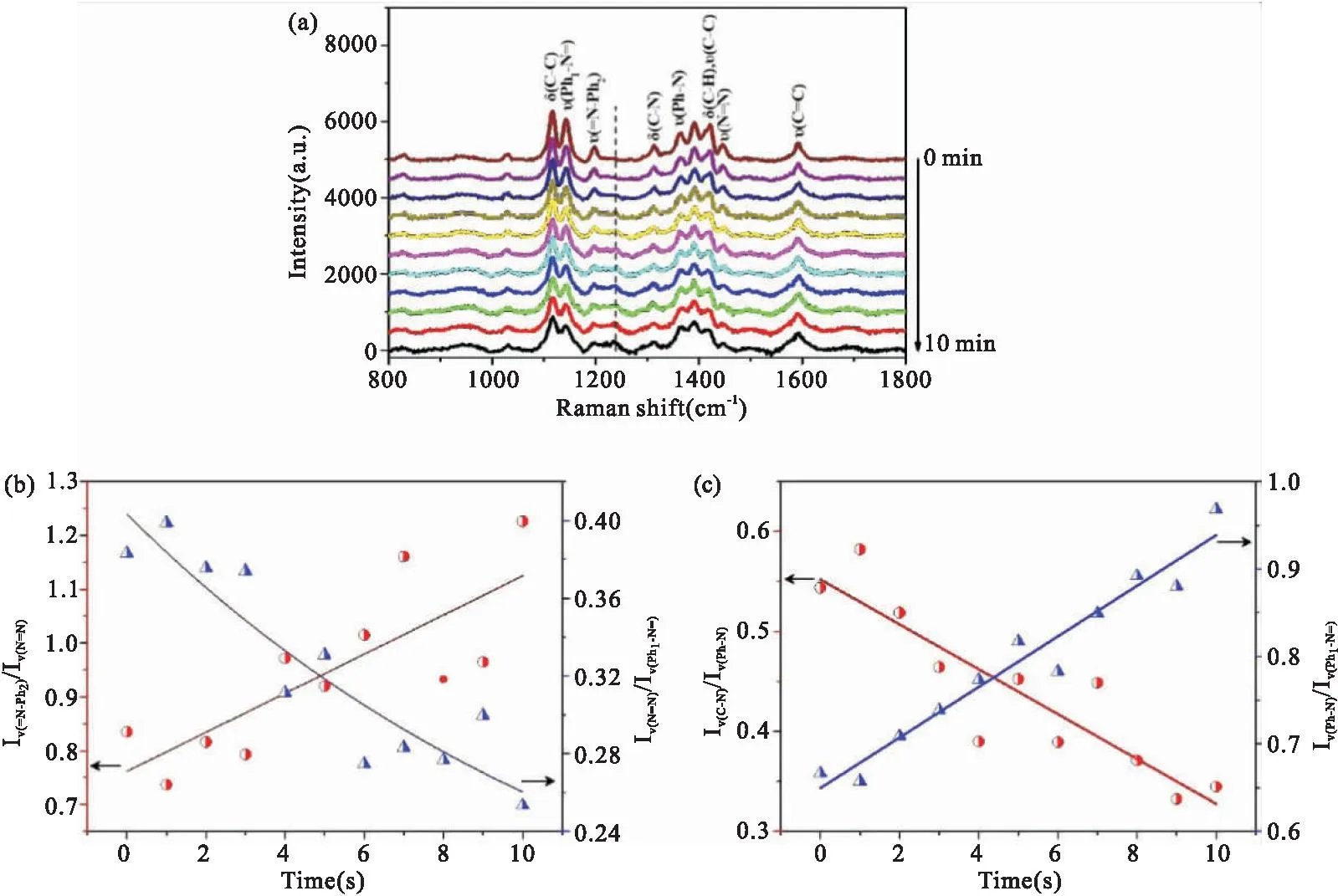
图6 Cu2O-Au表面MO随时间变化的SERS谱图(a),随时间变化的1197-1444 cm-1(圆形)和1444-1145 cm-1(三角形)处的拉曼谱峰强度比值(b)以及随时间变化的1313-1368 cm-1(圆形)和1368-1145 cm-1(三角形)处的拉曼谱峰强度比值(c)Fig.6 Time-dependent SERS spectra of MO adsorbed on Cu2O-Au(a), Time-dependent relative intensities of Raman bands at 1197 to 1444 cm-1 and 1444 to 1145 cm-1 (b) ; Time-dependent relative intensities of Raman bands at 1313 to 1368 cm-1 and 1368 to 1145 cm-1 (c)
为了进一步研究光催化降解MO过程中各化学键的断裂程度,对相关化学键对应的SERS谱峰峰强随激光照射时间的变化进行了分析。如图6b所示,随时间变化的1197-1444 cm-1处的SERS谱峰强度比值呈现上升趋势,而随时间变化的1444-1145 cm-1处的拉曼谱峰的强度比值呈现下降趋势,位于1145 cm-1、1197 cm-1处的峰分别归属于MO的υ(Ph1-N=)、υ(=N-Ph2)谱峰,位于1145 cm-1处的峰归属于MO的υ(N=N)谱峰,这说明不管是第一个苯环上的Ph1-N=还是第二个苯环上的=N-Ph2,其断裂程度均比N=N断裂程度低。相比于Ph1-N=、=N-Ph2,N=N对Cu2O-Au催化剂更为敏感,更容易断裂,这与常规拉曼测试结果中υ(N=N)最先消失的结果保持一致。
如图6c所示,随时间变化的1313-1368 cm-1处的SERS谱峰强度比值呈现下降趋势,而随时间变化的1368-1145 cm-1处的SERS谱峰强度比值呈现上升趋势,这说明三个化学键对催化剂敏感程度排序为Ph1-N=、C-N >Ph-N。因此,以Cu2O-Au作为催化剂,光照下MO分子中各化学键断裂程度从高到低依次为N=N >Ph1-N=、=N-Ph2, Ph1-N=、C-N >Ph-N。
4 总结
综上所述,制备了一种双功能复合材料Cu2O-Au,并将其用于甲基橙光催化降解过程的原位SERS研究。该复合材料相对于单一的Cu2O,具有良好的SERS以及光催化活性,主要因Au纳米粒子的SPR特性以及复合材料界面处肖特基势垒的存在对Cu2O催化性能的促进作用。结合复合材料的SERS和光催化活性对MO降解过程进行了在线监测,并进一步研究了其详细的分子机制。结果表明,在激光照射下,MO分子中各化学键断裂容易程度由高到低依次为N=N >Ph1-N=、=N-Ph2, Ph1-N=、C-N >Ph-N。MO分子中的N=N比Ph-N对Cu2O-Au催化剂更为敏感,更容易发生断裂,其中最难断裂为苯环上的C=C。此外,该双功能Cu2O-Au复合材料是一种有效的光催化剂和SERS基底,在光催化过程的原位SERS研究领域具有巨大的潜力。
猜你喜欢
探测与控制学报(2023年4期)2023-09-12 07:26:12
风流一代·经典文摘(2023年5期)2023-05-21 11:42:11
陶瓷学报(2021年3期)2021-07-22 01:05:06
分析科学学报(2021年3期)2021-07-14 01:51:16
色谱(2021年6期)2021-05-06 02:18:56
科技资讯(2020年12期)2020-06-03 04:44:20
新世纪智能(数学备考)(2019年9期)2019-10-16 11:44:58
电子测试(2018年18期)2018-11-14 02:30:36
物理学进展(2017年1期)2017-02-23 01:35:44
光学精密工程(2016年1期)2016-11-07 09:01:00
