
基于分子动力学模拟与实验验证探讨黄芩素抑制PD-1/PD-L1 相互作用的分子机制
2024-01-15 04:58:14郭怡琳刘柏平徐建国
食品工业科技 2024年2期
郭 妍,郭怡琳,刘柏平 ,徐建国,
(1.山西师范大学食品科学学院,山西太原 030031;2.华南农业大学材料与能源学院,广东广州 510630;3.华南农业大学教育部生物材料与能源重点实验室,广东广州 510630)
程序性细胞死亡-1(programmed cell death-1,PD-1)是一种重要的免疫检查点蛋白,可通过与配体PD-L1(programmed cell death ligand 1)结合抑制T细胞增殖,从而起到负向免疫调控作用以维持机体的免疫稳态[1]。然而肿瘤细胞能够通过过量表达PDL1 逃避机体的免疫监视,从而快速增殖、转移。因此阻断PD-1/PD-L1 通路可逆转机体的免疫抑制状态,有助于提高T 细胞对肿瘤细胞的杀伤力[2]。实际上,目前已有大量靶向PD-1 或PD-L1 的单抗抑制剂上市,且这些抑制剂在多种肿瘤临床治疗中取得了突破性进展[3],但单抗由于其较大的分子量而存在肿瘤渗透性差、稳定性低以及研发成本高等局限性[4-6]。小分子(MW<550 Da)抑制剂因可克服上述缺点而受到广泛关注。2015 年,百时美施贵宝公司报道了一系列抑制活性较好(IC50为0.92 nmol/L~14.25 μmol/L)合成小分子。之后,Zak 团队阐明了该类小分子的抑制机制,指出小分子可作用于PDL1 表面并诱导PD-L1 的二聚最终结合到二聚体的内表面,从而阻断该通路[7-8]。
相比之下,天然的小分子抑制剂具有生物活性多样、毒副作用低和来源广泛等特点,为该通路抑制剂的发现提供了巨大宝库[9]。黄芩素是一种天然黄酮类化合物,已有研究报道其可通过调控PD-L1 表达抑制PD-1/PD-L1 水平[10-11],然而关于其能否直接抑制PD-1/PD-L1 相互作用及分子机制的研究鲜有报道。分子动力学模拟是利用计算机软件,以经典力学为基础、分子随时间变化的动态行为为模拟对象的一种方法,是研究分子间相互作用的重要工具[12]。事实上,分子动力学模拟已成功应用于PD-1/PDL1 通路的研究。如Sun 等[13]和Shi 等[14]利用分子动力学模拟的方法研究了PD-L1 与单抗的作用机制,发现PD-L1 的CC'FG 结构域为主要结合区域。Verdura 等[15]通过分子动力学模拟发现白藜芦醇可直接结合到PD-L1 dimer 内表面从而阻断PD-1/PDL1 相互作用。此外,本课题组前期的分子动力学模拟研究表明姜黄素等天然小分子能够直接与PD-L1 dimer 的关键残基结合从而抑制该通路[16-18]。
本文首先利用分子对接构建PD-L1 dimer/黄芩素复合物体系,然后借助GROMACS 2016.4 软件进行150 ns 的分子动力学模拟。通过分析结合自由能、接触数和非键相互作用等获得黄芩素与PD-L1 dimer 的结合亲和力、作用位点及作用类型等信息,从理论层面掌握其直接抑制PD-1/PD-L1 相互作用的分子机制。最后利用酶联免疫吸附试验(Enzyme-Linked Immunosorbent Assay,ELISA)对黄芩素的抑制作用进行验证,这些数据有助于指导该通路天然小分子抑制剂的发现。
1 材料与方法
1.1 材料与仪器
黄芩素(纯度≥98%,HPLC)上海源叶生物科技有限公司;PD-1 酶联免疫分析试剂盒、人源PD-1标准品 本生(天津)健康科技有限公司。
SHJ-4A 数显恒温磁力搅拌水浴锅 常州国语仪器制造有限公司;Multiskan FC 型酶标仪 赛默飞世尔(上海)仪器有限公司。
1.2 实验方法
1.2.1 体系构建及分子对接 根据文献[7-8,15-16,18]调研结果,发现小分子抑制剂直接抑制PD-1/PD-L1 相互作用的关键在于PD-L1 dimer 的形成与稳定。基于此,首先从PDB 数据库下载PD-L1 dimer/BMS-200(PDB ID:5N2F)和黄芩素(PDB ID:4X2A)的结构。删除水分子和配体BMS-200 得到PD-L1 dimer,并运用WHAT IF 服务器修复其缺失部分,黄芩素则在Chem3D 软件的MM2 力场下进行能量最小化,结构见图1。

图1 PD-L1 dimer(a)和黄芩素(b)的分子结构Fig.1 Structures of the PD-L1 dimer (a) and baicalein (b)
然后利用AutoDock Tools 分别对PD-L1 dimer和黄芩素进行加氢处理并生成pdbqt 文件,使用Grid Box 模块将盒子大小设为20 Å×20 Å×20 Å,格点间距设为1 Å,其他参数均设为默认值,最后运行AutoDock Vina 软件进行分子对接[19]。选择结合亲和力最强的对接构象作为分子动力学模拟的初始结构。运用PyMOL 软件可视化对接结果,构建相互作用3D 模式图。为了考察黄芩素对PD-L1 dimer 构象的影响,分别构建含黄芩素和不含黄芩素的体系。为了简便,将两个体系分别称为复合物和Dimer,PD-L1 dimer 的两条链则分别命名为APD-L1 和BPD-L1。
1.2.2 分子动力学模拟 使用GROMACS 2016.4软件进行分子动力学模拟,分别采用Amber ff99SB[20]和GAFF[21]描述蛋白和小分子的力场参数。首先将黄芩素和Dimer 体系置于立方体的溶剂盒子,并保证体系原子到盒子边缘的距离不小于10 Å,随后添加抗衡离子以保证体系呈电中性。紧接着采用最陡下降法和共轭梯度法对各体系进行能量最小化,以使体系达到收敛状态。然后依次进行1 ns 的NVT 和1 ns 的NPT 平衡,使用V-rescale 耦合方法将温度维持在300 K,使用Parrinello-Rahma 方法将压力维持在1 atm。最后,体系在相同的参数下进行150 ns 的模拟,并将快照以1.0 ps 为时间间隔保存至轨迹用于后续分析。模拟过程中,短程非键相互作用的截断距离设为10 Å,长程静电相互作用的计算采用Particle Mesh Ewald(PME)算法,氢原子的约束使用LINCS算法。
1.2.3 结合自由能 采用分子力学泊松玻尔兹曼表面积法(molecular mechanics Poisson-Boltzmann surface area,MM-PBSA)计算各体系的结合自由能[22]。从动力学最后30 ns 的平衡轨迹中采集300 帧构象用于结合自由能(ΔG)计算,公式如下:
式中,Gcomplex、Gprotein和Gligand分别代表蛋白-配体复合物、蛋白和配体的自由能;EMM代表气相中的分子力学能,包括范德华相互作用能(Evdw)和静电相互作用能(Eele);Gsol代表溶解自由能,可分解为极性溶解自由能(EPB)和非极性溶解自由能(ESA)两部分。前者通过求解泊松-玻尔兹曼(Poisson-Boltzmann,PB)方程获得,后者则通过计算分子表面面积(surface area,SA)而得,TΔS 为熵变对结合自由能的贡献。由于本文关注结合自由能的相对值,且熵变的计算耗时较长,因此不考虑熵变的影响。为了解黄芩素与PD-L1 dimer 作用过程中的关键残基,本文采用与前期研究相同的方法对结合自由能进行了分解[22-23]。
1.2.4 轨迹分析 使用GROMACS 2016.4 内置分析工具和分子可视化程序1.9.3(Visual Molecular Dynamics,VMD)对模拟轨迹进行分析。使用mindist 工具计算复合物体系的接触数,截断值设为6.0 Å。蛋白在模拟过程中二级结构的变化利用dictionary of secondary structure of proteins(DSSP 2.0.4)工具进行分析。采用主成分分析(principal component analysis,PCA)方法描述蛋白原子运动的相关性[24]。利用VMD 程序计算复合物体系分子间氢键占有率[25],其中氢键的判定标准为受体-氢-供体的角度>135°且受体-氢原子的距离<3.5 Å。
1.2.5 ELISA 通过使用PD-1 ELISA 试剂盒测定反应体系中PD-1 含量,计算黄芩素对PD-1/PD-L1相互作用的抑制率。具体操作步骤包括加样:向空白孔、标准孔和待测样品孔分别加入二甲基亚砜、标准品和待测样品50 μL,37 ℃反应30 min;加酶:用洗涤液洗板5 次,除空白孔外每孔加入酶标试剂50 μL,37 ℃反应30 min;显色:用洗涤液洗板5 次,依次加入显色剂A 和显色剂B 50 μL,37 ℃避光反应10 min;终止:加入终止液50 μL。最后以空白孔调零,450 nm波长处测定各孔的吸光度。以标准品浓度为横坐标,吸光度为纵坐标,绘制标准曲线y=0.029x-0.0161,R²=0.997,标准品在0~60 ng/L 范围内线性良好。
1.3 数据处理
所有试验均重复测定三次或以上,使用Microsoft Office Excel 2010 软件进行绘图,采用SPSS 27 软件进行统计分析,P<0.05 代表差异显著,试验结果表示为平均值±标准差。
2 结果与分析
2.1 均方根偏差分析
通过计算黄芩素周围20 Å范围内残基的均方根偏差(root mean square deviation,RMSD)值,可以考察体系在150 ns 分子动力学模拟过程中的稳定性。对于Dimer 体系,选择与复合物体系相同的残基并计算其RMSD 值。由图2 可知,两个体系的RMSD值变化情况有所不同,复合物体系在3 ns 内急速上升,之后波动幅度较小,随着模拟时间的延长,稳定在2.66 Å左右。Dimer 体系在5 ns 时突增至3.07 Å,5~30 ns 波动在3.50 Å左右,但30 ns 后,RMSD波动幅度较大,甚至高达5.35 Å,60 ns 后RMSD 值基本收敛至3.11 Å。说明复合物比Dimer 体系具有更高的结构稳定性。综上,两个体系均达到了平衡,可用于后续分析。

图2 复合物和Dimer 体系在模拟过程中的均方根偏差Fig.2 Root mean square deviation results of the complex and Dimer systems during simulations
2.2 均方根波动分析
为了比较复合物和Dimer 体系在模拟过程中构象柔性的差异,计算了PD-L1 dimer 残基的均方根波动(root mean square fluctuation,RMSF)值,结果见图3。整体来看,除少数区域外,两个体系RMSF 的趋势较为相似,但复合物比Dimer 体系具有更低的柔性。进一步观察发现波动较大的残基主要位于两个体系的N 端、C 端和loop 区,其中Dimer 体系BC loop 区残基RMSF 值达到了5 Å。复合物体系结构柔性显著较低的区域主要集中于β折叠结构域,其RMSF 值仅为1~3 Å,如C-sheet(残基54~59)、F-sheet(残基121~124)和G-sheet(残基110~117)。综上,黄芩素的结合主要降低了sheet 区域的柔性,重要的是该区域为PD-1 与PD-L1 的主要结合位点,这与以往的研究结果相似[7-8,15-16,18]。
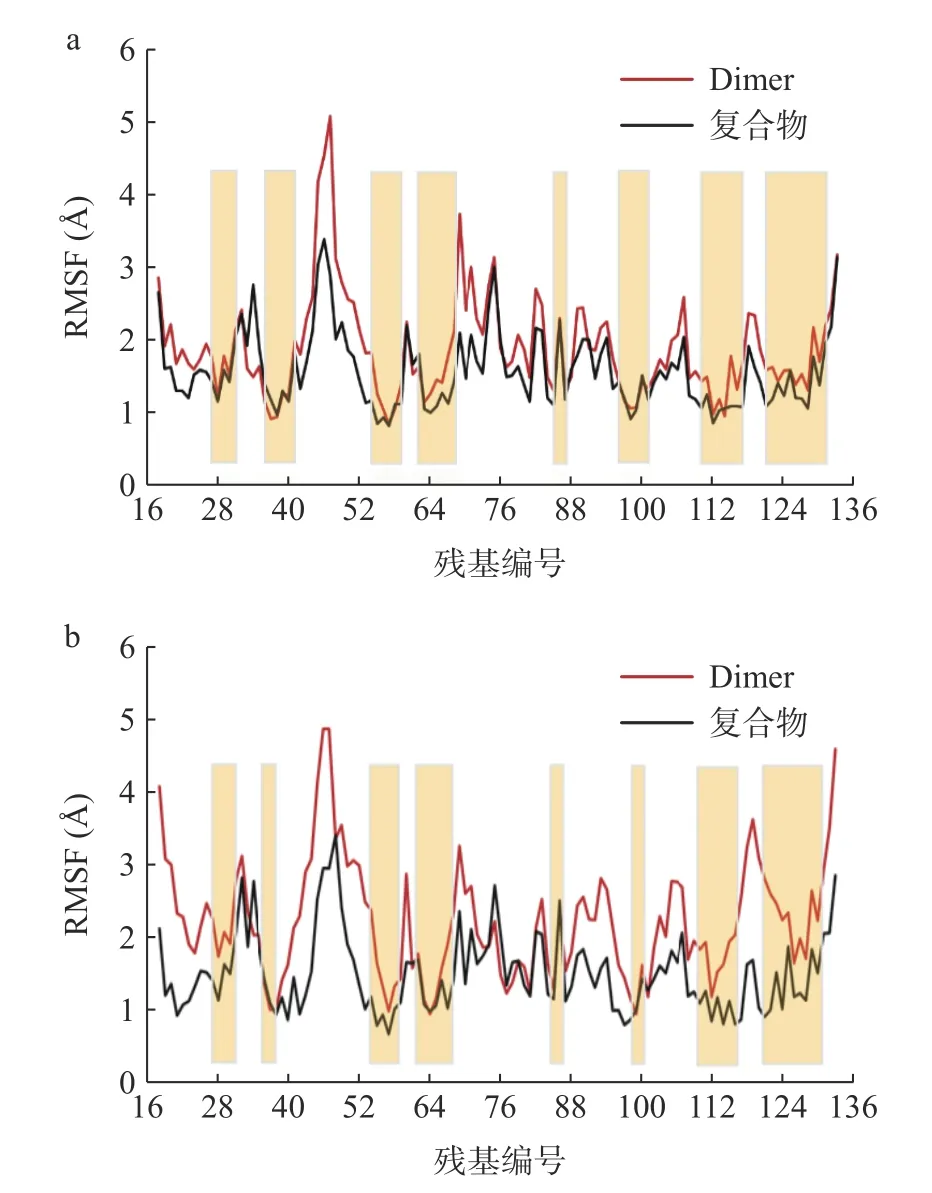
图3 模拟过程中APD-L1(a)和BPD-L1(b)残基的均方根波动Fig.3 Root mean square fluctuation of each residue on theAPD-L1 (a) andBPD-L1 (b) during simulations
2.3 结合自由能分析
抑制剂与蛋白的结合自由能可用于描述两者间结合亲和力的强弱[26]。为了评估这一特性,本文利用g_mmpbsa 程序计算了黄芩素与PD-L1 dimer 的结合自由能,并详细解析了相互作用成分的能量贡献。由表1 可知,黄芩素与PD-L1 dimer 的ΔG 为-32.41±0.31 kcal/mol,说明两者具有较强的结合亲和力。相比之下,Dimer 体系的ΔG 为正值(36.11±0.89 kcal/mol),说明黄芩素是PD-L1 dimer 稳定存在的必备条件。根据MM-PBSA 方法,ΔG 可表示为总极性能(ΔEpolar,total)和总非极性能(ΔEnonpolar,total)之和,两者可进一步分解为ΔEele+ΔEPB和ΔEvdw+ΔESA。复合物体系的ΔEvdw和ΔESA分别为-45.61±0.19和-3.41±0.03 kcal/mol,说明两者均有利于黄芩素的结合。ΔEele也提供了-2.61±0.47 kcal/mol 的有利贡献,但完全被不利于黄芩素结合的极性溶解自由能(19.22±0.60 kcal/mol)抵消,产生了不利于结合的总极性相互作用(16.61±1.07 kcal/mol)。综上所述,总非极性相互作用是黄芩素与PD-L1 dimer 结合的主要驱动力,而总极性作用不利于结合,这类似于本人前期关于天然小分子抑制PD-1/PD-L1 相互作用的研究[23]。此外,黄芩素抑制该通路的能力近似于姜黄素,但强于白藜芦醇和表没食子儿茶素没食子酸酯[23]。
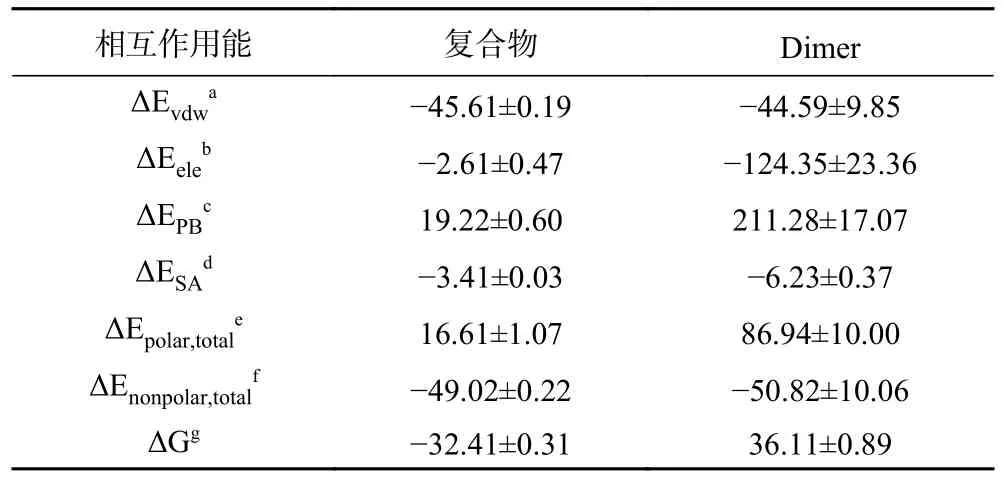
表1 复合物和Dimer 体系的结合自由能(kcal/mol)Table 1 Binding free energies of the complex and Dimer systems (kcal/mol)
2.4 结合能分解
使用MM-PBSA 方法对总结合自由能进行分解以识别对黄芩素结合有重要作用的残基,结果见图4。黄芩素与PD-L1 dimer 的6 个残基产生了强于-1 kcal/mol 的相互作用,分别是ATyr56、AMet115、AAla121、BMet115、BAla121 和BAsp122。在这些残基中,AMet115 与黄芩素的相互作用最强,作用能为-2.34 kcal/mol;ATyr56 次之,结合自由能贡献为-1.49 kcal/mol;BAsp122 的贡献最小,为-1.03 kcal/mol。此外,其他残基如AIle54 和AIle116 对黄芩素的结合也起到了一定的作用,两者的贡献分别为-0.91 和-0.90 kcal/mol。综上所述,Tyr56、Met115、Ala121 和Asp122 为复合物体系的关键残基。本人前期的研究表明Tyr56 和Met115 为天然小分子姜黄素、白藜芦醇和表没食子儿茶素没食子酸酯的关键结合残基。类似地,Zak 等[7]和Ahmed 等[27]的研究表明,残基Met115 和Tyr56 侧链结构重排有利于合成小分子结合。重要的是这些关键残基也参与了PD-L1/PD-1 相互作用。综上,从能量方面看,黄芩素可直接与PD-L1 dimer 稳定结合。

图4 复合物体系的残基自由能分解Fig.4 Residue energy decomposition of the complex system
2.5 接触数分析
为了确定黄芩素结合区域,计算了复合物体系的分子间接触数,并将与黄芩素接触数大于10 的残基视为重要残基[28]。由图5 可知,黄芩素主要结合在PD-L1 dimer 的4 个区域,分别为C-sheet(残基54~56)、C'-sheet(残基66~68)、F-sheet(残基115~117)和G-sheet(残基121~124)。其中,C-sheet 的残基Ile54 和Tyr56 与黄芩素有很大的接触数分布,Val55与黄芩素的接触稍弱,但AVal55 的接触数大于10。因此这三个残基为该区域的重要残基。C'-sheet 三个残基与黄芩素的接触较弱,其中极性残基AGln66 与黄芩素之间的接触数最大,这主要来源于氢键的贡献(图6)。F-sheet 残基与黄芩素的接触数分布较大,其中非极性残基AMet115 与黄芩素的接触数为34。G-sheet 非极性残基AAla121、BAla121 和极性残基BAsp122 分别贡献了30、36 和23 的接触数,这与结合能分解结果一致。表2 为黄芩素与结合区域的接触数。由表可知,复合物体系接触数总和为389,这与姜黄素的相关结果类似[23]。此外,黄芩素与各结合区域的接触数呈G-sheet>F-sheet>Csheet>C'-sheet 趋势,说明黄芩素与G-sheet 的结合最强,而与C'-sheet 的结合最弱。

表2 黄芩素与结合区域的接触数Table 2 Contact numbers between baicalein and the binding domains

图5 黄芩素与PD-L1 dimer 之间的接触数Fig.5 Contact numbers of baicalein with the PD-L1 dimer

图6 复合物体系的详细结合模式Fig.6 Detailed binding mode of the complex system
2.6 非键相互作用分析
基于黄芩素的结构,猜测其与PD-L1 dimer 之间的相互作用类型主要为氢键和疏水相互作用。因此首先对复合物体系分子间氢键占有率进行了定量分析。如表3 所示,黄芩素O2 和O3 原子分别与残基AVal55 和AGln66 形成占有率为15.28%和10.30%的氢键。表明氢键在模拟过程中不稳定,这与静电相互作用能结果一致(-2.61 kcal/mol)。然后对复合物体系的结合模式进行了评价。如图6 所示,黄芩素位于APD-L1 和BPD-L1 之间圆柱形结合口袋中,且被残基BIle54、AMet115、AAla121、BMet115、BAla121、ATyr56、BAsp122、AVal55 和AGln66 的侧链包围,其中前5 个残基与黄芩素形成了紧密的疏水相互作用。这与结合能分解结果一致地表明,残基Tyr56、Met115、Ala121 和Asp122 可能是PD-1/PD-L1 通路天然的小分子抑制剂的潜在靶点。此外,相比于氢键,黄芩素的结合更依赖于苯环与疏水残基之间的疏水相互作用。

表3 复合物体系的氢键占有率Table 3 Hydrogen bond occupancies of the complex system
2.7 互相关矩阵分析
为了探究黄芩素对PD-L1 dimer 原子运动性的影响,计算了残基Cα原子的互相关矩阵[29]。矩阵中对角线颜色表示残基的运动程度,对角线外红色部分(正值)代表残基间运动呈正相关,即残基间的动力学运动方向一致。而蓝色部分(负值)代表残基间的运动有负相关性,即残基间的动力学运动方向相反[30]。如图7 所示,对角线尤其是残基132~143 和242~250(分别对应于APD-L1 的132-143 和BPD-L1 的132-141)颜色偏红,说明两个体系C 端残基均具有较高的灵活性。此外,复合物体系蓝色区域较少,而Dimer 体系蓝色区域占据了矩阵大部分面积,说明该体系中APD-L1 和BPD-L1 之间残基运动具有更强的负相关性。综上,黄芩素可稳定PD-L1 dimer 的结构。

图7 复合物(a)和Dimer 体系(b)Cα 原子的互相关矩阵Fig.7 Cross-correlation matrixes for Cα atoms in the complex(a) and Dimer systems (b)
2.8 二级结构分析
利用DSSP 程序计算了黄芩素和Dimer 体系蛋白二级结构随时间的变化规律。如图8 所示,Dimer体系的β-折叠,如残基37~42、93~100 和104~114等(分别对应于APD-L1 的残基54~59、110~117 和121~123)稳定存在于整个动力学模拟过程。残基32~35(APD-L1 的残基49~52)主要在3-螺旋和转角之间相互转变,其中3-螺旋出现的更频繁,而残基158~161(BPD-L1 的残基49~52)表现出相反的动力学行为;残基72~77 和198~204(分别对应于APDL1 的残基89~94 和BPD-L1 的残基89~95)的二级结构包括3-螺旋、α-螺旋和转角;残基116~126(APD-L1 的残基133~143)的二级结构在前84 ns 表现为α-螺旋,之后转变为转角、3-螺旋和无规则卷曲;残基242~249(BPD-L1 的残基133~140)也包括这些二级结构,但这些构象时而出现时而消失,表明PD-L1 dimer 的C 端不稳定。复合物体系二级结构的组成类似于Dimer 体系,整个模拟过程中β-折叠构象一直保持稳定,其中C-sheet、F-sheet 和Gsheet 是PD-1 与PD-L1 的关键结合区域。因此可以推断黄芩素能与PD-L1 dimer 稳定结合从而抑制PD-1/PD-L1 相互作用。
2.9 PD-1/PD-L1 相互作用抑制率
为了阐明黄芩素对PD-1/PD-L1 相互作用的抑制能力,以BMS-202 为阳性对照进行了ELISA 试验,结果见图9。由图9 可知,随着BMS-202 和黄芩素浓度的增加,两者对PD-1/PD-L1 相互作用的抑制率呈显著升高趋势(P<0.05),且BMS-202 的抑制率始终大于黄芩素。当浓度为6.25 μg/L 时,黄芩素对PD-1/PD-L1 通路的抑制率(17.95%±0.92%)与BMS-202 的抑制率(19.69%±0.85%)差异不显著(P>0.05)。当浓度为50 μg/L 时,黄芩素与BMS-202 的抑制率分别达到43.43%和52.06%,差异显著(P<0.05)。黄芩素与BMS-202 对PD-1/PD-L1 相互作用抑制率的IC50分别为79.47 和44.29 μg/L,表明黄芩素对该通路较强的抑制作用[31-32]。
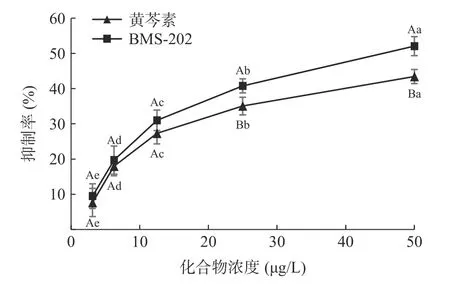
图9 黄芩素对PD-1/PD-L1 相互作用的抑制率Fig.9 Inhibitory rate of baicalein on the PD-1/PD-L1 interaction
3 结论
本文利用分子动力学模拟和实验验证研究了黄芩素抑制PD-1/PD-L1 相互作用的分子机制。构象动力学结果表明,PD-L1 dimer 与黄芩素的结合在整个模拟过程中基本保持稳定;结合自由能结果表明,黄芩素稳定PD-L1 dimer 的能力较强为-32.41±0.31 kcal/mol;通过对结合能进行分解,发现PD-L1 dimer 的4 个关键残基Tyr56、Metl15、Ala121 和Asp122 对黄芩素结合有重要贡献;结合模式和相互作用结果表明黄芩素主要结合在PD-L1 dimer 的Csheet、F-sheet 和G-sheet 区域,其中苯环与关键残基间的非极性相互作用是复合物稳定的主要因素;互相关矩阵和二级结构结果进一步表明,黄芩素可稳定结合在sheet 区域。ELISA 结果表明黄芩素是PD-1/PD-L1 相互作用的有效阻断剂。总之,这些信息有助于筛选PD-1/PD-L1 通路天然小分子抑制剂。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
时代英语·高一(2019年5期)2019-09-03 02:09:34
中成药(2018年7期)2018-08-04 06:04:18
中成药(2018年3期)2018-05-07 13:34:18
电测与仪表(2016年11期)2016-04-11 12:20:42
池州学院学报(2015年3期)2016-01-05 01:13:04
电源技术(2015年5期)2015-08-22 11:18:28
天津科技大学学报(2015年2期)2015-08-09 01:40:42
