
戊二酸锌体系催化合成二氧化碳基聚碳酸酯研究进展
2024-01-04 12:31:54吴玉龙聂迎芳奚桢浩
分子催化 2023年5期
刘 振, 吴玉龙, 聂迎芳, 孙 莉, 奚桢浩
(华东理工大学 化学工程联合国家重点实验室; 上海市多相结构材料化学工程重点实验室, 上海 200237)
CO2是一种无毒无害、 来源丰富、 可再生的碳一资源[1-4].CO2的化学转化是目前研究的热点之一, 受到化学工作者的广泛关注[5-8].CO2的化学转化主要包括:1) 羧化、环化偶联反应; 2) 光电还原反应; 3) 氢化反应; 4) 合成丙烯酸酯/盐; 5) 合成聚碳酸酯等.其中, CO2和环氧化物开环共聚合成的聚碳酸丙烯酯(PPC)[9], 可以用做生物降解的地膜材料,从而代替不可降解的低密度聚乙烯地膜, 有着较大的市场应用前景.
由于CO2固有的化学稳定性, 以戊二酸锌为代表的催化剂体系是CO2化学转化合成PPC 的关键技术[10].1969 年Inoue 等[11]使用二乙基锌(ZnEt2)/水作为催化剂, 首次以CO2为原料合成了PPC, 活性为13.4 g聚合物/g催化剂(gpoly/gcat).此后, 研究者们开发了众多基于锌和其他金属的非均相催化剂, 主要包括戊二酸锌(ZnGA)催化剂[12]、双金属氰化物(DMC)催化剂[13]、稀土配合物催化剂[14]、金属卟啉催化剂[15]、Salen 催化剂[16]等.其中ZnGA 由于合成过程简单、原料廉价易得, 是一类研究较为广泛的CO2和环氧化物共聚的催化剂体系[9].我们从戊二酸锌催化剂, CO2/PO 二元共聚以及CO2/PO/第三单体三元共聚等方面进行综述, 旨在阐述ZnGA 催化剂在CO2基聚碳酸酯合成领域的研究进展.
1 戊二酸锌催化剂
传统ZnGA 催化剂具有制备简单、无毒、廉价和副产物含量低等优点, 是CO2和环氧丙烷(PO)共聚的非均相催化体系之一[17-19].催化剂的活性中心及配体环境对CO2/PO 共聚反应有显著影响.2003 年, Ree 课题组[20]发现ZnGA 表面的羧基基团为层状结构.与外层结构相比, 内层结构中含有更多的Zn-OH 活性基团.PO 分子比CO2更容易吸附在ZnGA 表面上, 并插入Zn—O 键, 这表明ZnGA 催化CO2/PO 的共聚反应由PO 分子的配位引发(图1).随后Kim 等[21]采用单晶X 射线, 再次证实了ZnGA 的层状结构.每个锌中心由不同位置的戊二酸基团的四个氧原子四面体配位, 从而形成锌和戊二酸离子的交替层状结构.
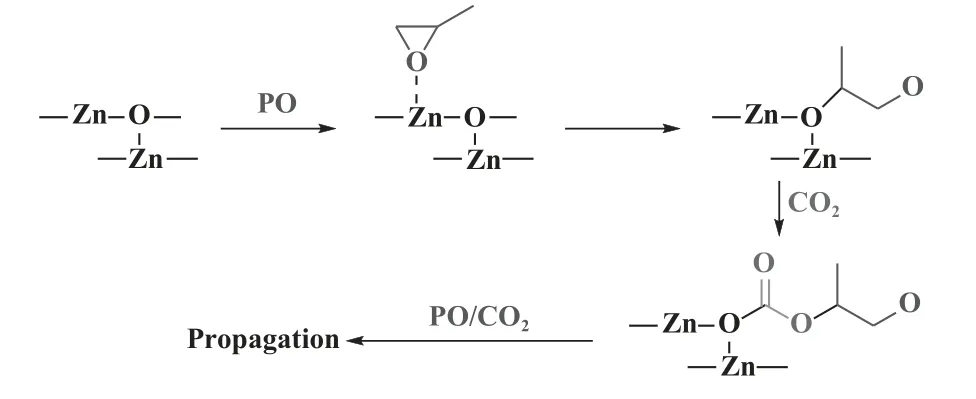
图1 ZnGA 催化CO2/PO 共聚引发机理[21]Fig.1 Proposed initiation mechanism for the copolymerization of PO and CO2 catalyzed by ZnGA[21]
戊二酸锌单晶结构的制备和表征对于研究其催化性能具有重要意义.2004 年, Ree 课题组[21]利用六水合高氯酸锌[Zn(ClO4)2·6H2O]和戊二腈[NC(CH2)3CN]首次合成了单晶sc-ZnGA(图2).Chisholm 等[22]同样认为CO2/PO 共聚发生在ZnGA 表面上的Zn-OH 活性位点.2020 年, Vineetha 等[23]首次采用凝胶扩散法生长了戊二酸锌晶体.热重分析(TGA)和差热分析(DTA)的结果表明, ZnGA 在410 ℃以下具有热稳定性, 在410~509 ℃分解为碳酸锌.
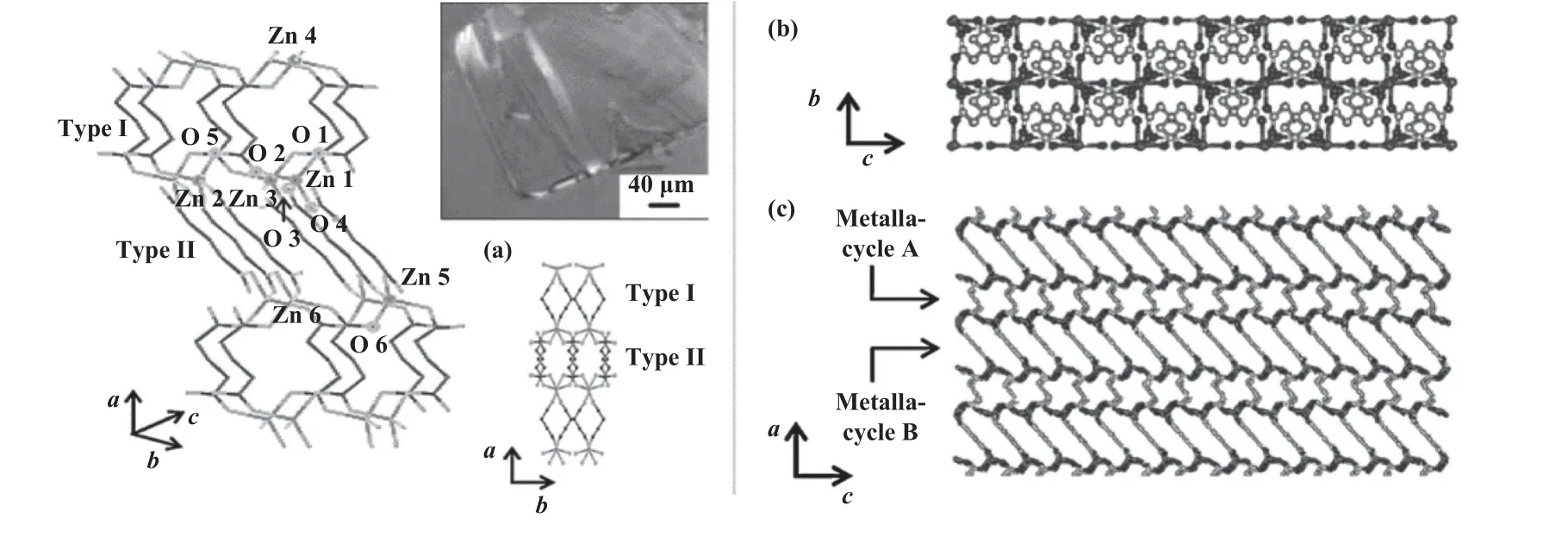
图2 ZnGA 单晶结构[21]Fig.2 The single crystal structure of ZnGA[21]
2 ZnGA 催化CO2/PO 二元共聚
2.1 锌源的影响
ZnGA 催化剂体系具有合成简单, 原料廉价易得, 产物PPC 分子量高、 副产物环状碳酸酯(CPC)少等特点.自Ionue 首次以CO2为原料合成PPC 后,科学工作者广泛研究了CO2和环氧化物开环共聚的催化体系.1999 年, Ree 课题组[24]考察了锌源对合成催化剂体系的影响.在氧化锌、氢氧化锌、硝酸锌和二乙基锌4 种锌源中, 以氧化锌(ZnO)为锌源合成的ZnGA 具有相对较低的表面积、较高的结晶度, ZnGA 的收率大于98%, 最高活性为70.0 gpoly/gcat, 产物PPC 的Mn为210.0 kDa.除ZnEt2外,氧化锌[24]、乙酸锌[25]、硝酸锌[26]、氢氧化锌[25]等均可作为锌源.2003 年, Ree 课题组[26]分别研究了以氧化锌、硝酸锌和二乙基锌合成的ZnGA 在CO2/PO 共聚反应中的催化活性.结果表明, 比表面积低, 但结晶度和结晶质量高的ZnGA 催化剂显示出更高的催化活性.ZnGA 催化剂中的锌原子与戊二酸配体的羧基氧以四面体的方式进行配位.催化剂的锌-氧距离为0.195~0.196 nm, 相邻的锌原子之间的距离为0.319~0.323 nm[25,27].
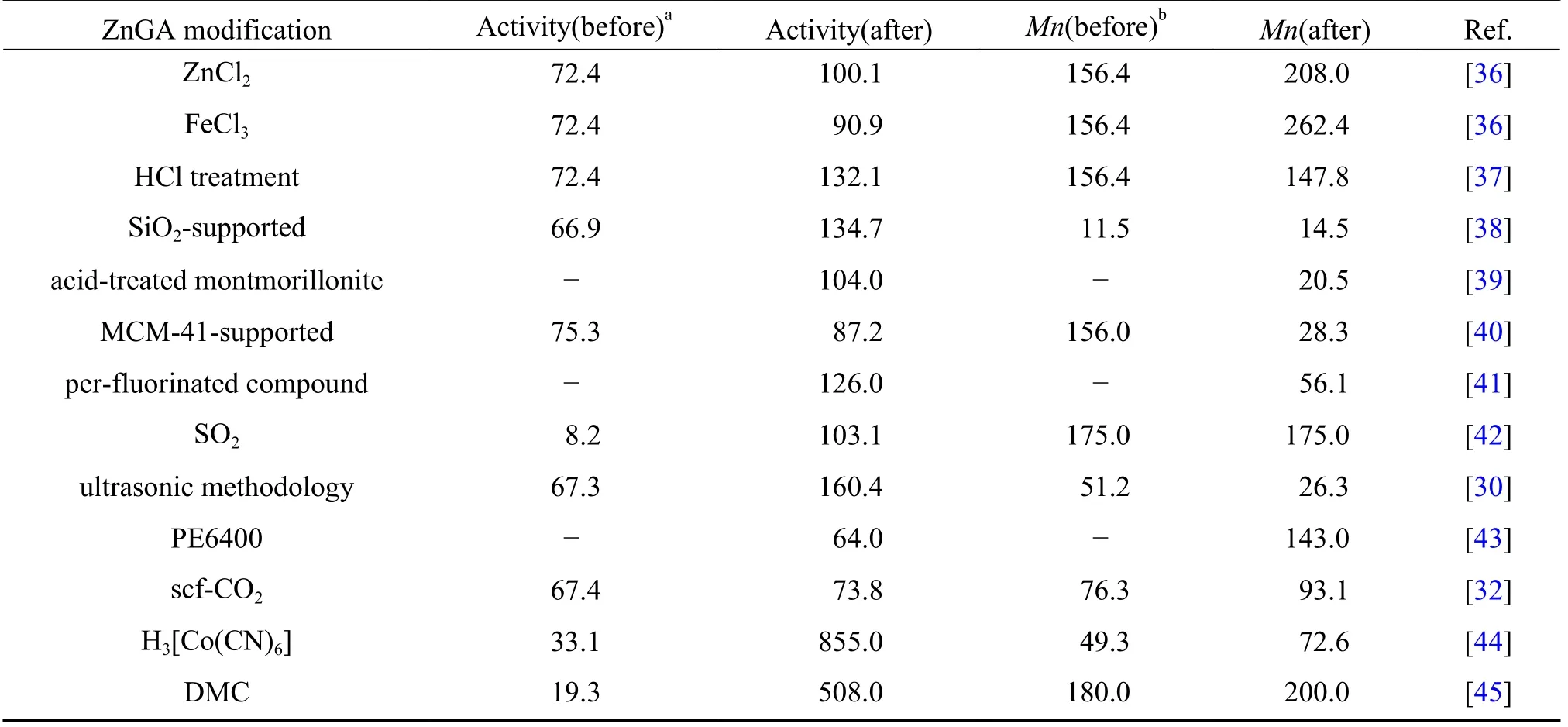
表1 改性ZnGA 催化CO2/PO 共聚反应Table 1 Copolymerization of CO2/PO catalyzed by modified ZnGA
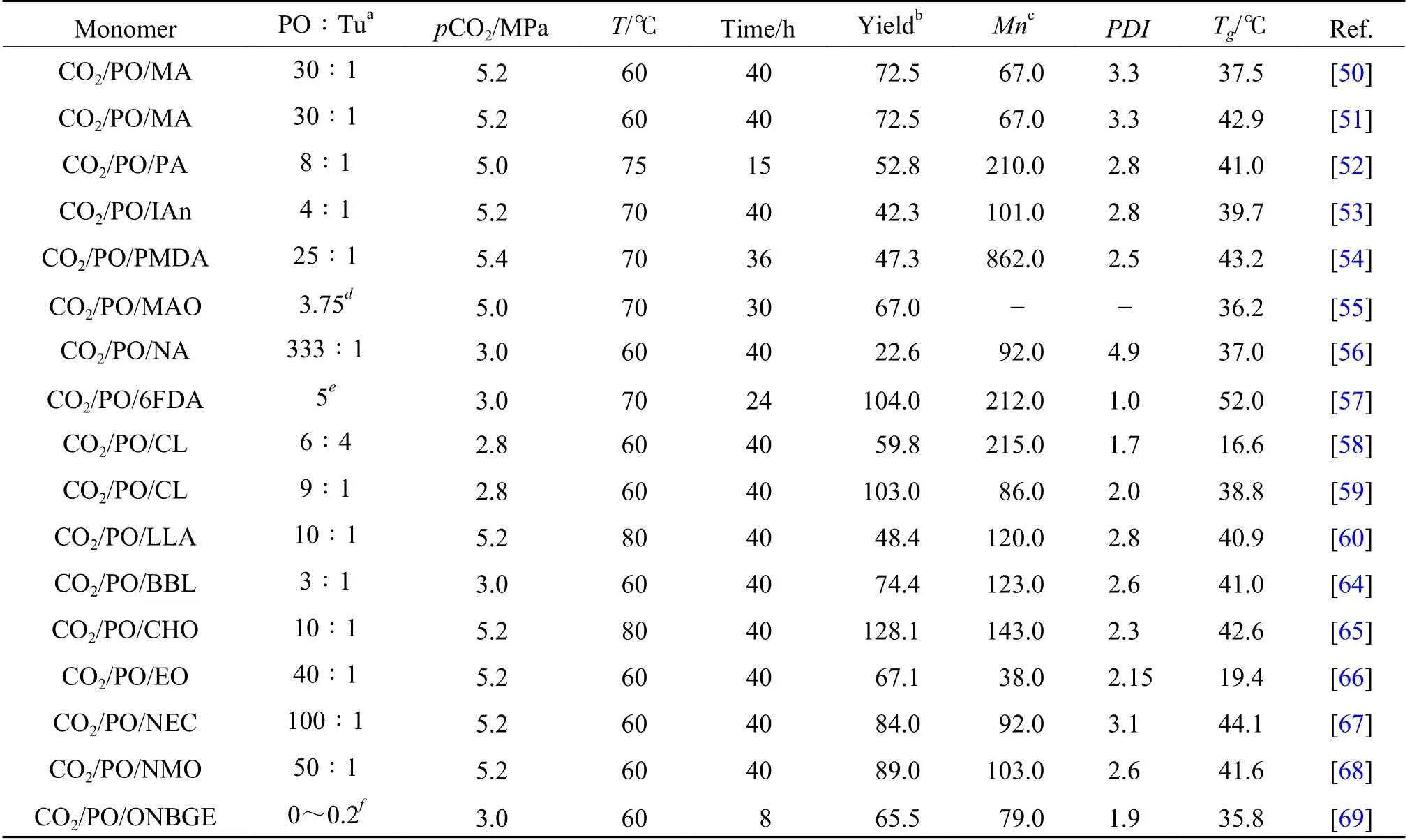
表2 不同条件下戊二酸锌催化的三元共聚反应Table 2 The terpolymerization reaction catalyzed by zinc dicarboxylate
2006 年, Ree 课题组[25]以不同的锌源合成了ZnGA(图3), 研究了ZnGA 催化剂的催化活性与结晶度和结晶质量的关系.结果表明, 以二水合乙酸锌(Zn(OAc)2·2H2O)合成的ZnGA 催化剂结晶度为77.6%, 具有花瓣般的形态结构和较大的表面积(48.5 m2/g), 其催化活性为83.0 gpoly/gcat, 产物分子量为160.0 kDa.当ZnGA 催化剂的结晶度和结晶质量相同时, 比表面积越大, 催化活性越高.
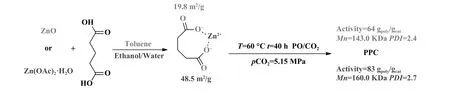
图3 不同锌源合成的ZnGA 催化CO2/PO 共聚反应[25]Fig.3 Copolymerization of PO and CO2 catalyzed by ZnGA from different Zn sources[25]
2.2 酸源的影响
1981 年, Soga 课题组[12]研究了一系列脂肪族二羧酸与氢氧化锌合成的催化剂体系, 结果表明,ZnGA 表现出最高的CO2/PO 共聚活性.Ree 课题组[25,28]使用11 种不同戊二酸衍生物与氧化锌反应(图4(a)).与ZnGA 相比(图4(b)), 采用戊二酸衍生物制备的催化剂在产率、催化活性和产物PPC 分子量等方面相对较低.其原因主要表现在以下两个方面:(1) 戊二酸中给电子基团或吸电子基团的引入改变了羧酸的亲和性, 从而导致Zn 金属中心路易斯酸度的变化; (2) 在ZnGA 周围引入侧基取代基增加了Zn 金属中心的空间位阻, 改变了羧酸盐与Zn 金属中心的配位特性, 降低了催化活性.2011 年,Rieger 课题组[29]对ZnGA 进行了实验表征和晶体结构的计算, 比较了琥珀酸锌、戊二酸锌、己二酸锌和庚二酸锌的催化特性.结果表明, 戊二酸锌、己二酸锌和庚二酸锌的双锌中心Zn—Zn 的距离为0.43~0.50 nm, 而琥珀酸锌的Zn—Zn 距离为0.48~0.68 nm.作者认为, Zn—Zn 间距离的不同是引起琥珀酸锌与同系物之间催化活性差异的主要原因.
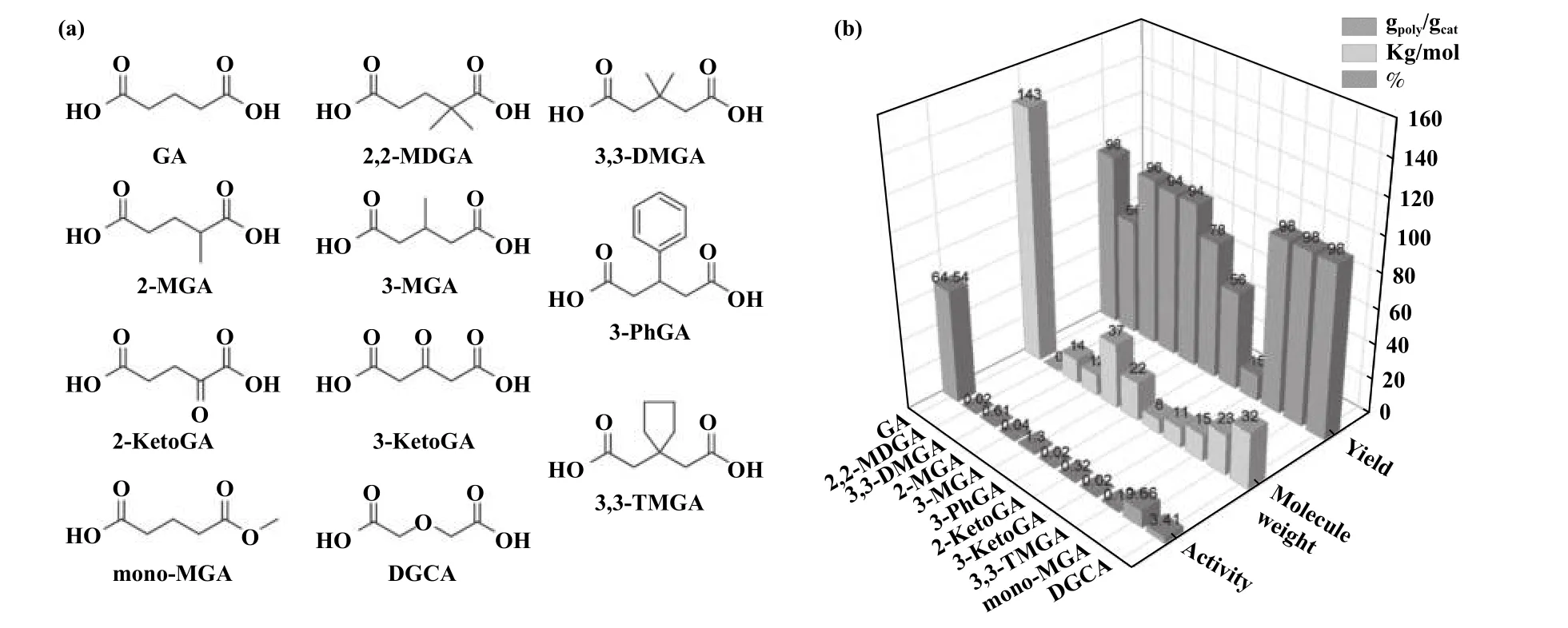
图4 (a) 戊二酸及其衍生物结构[25,29]; (b) 戊二酸衍生物对产率、 活性和分子量的影响Fig.4 (a) Structures of glutaric acid and its derivatives[25,29]; (b) The effect of different glutaric acid derivatives on yield, activity and molecular weight
2.3 制备过程的影响
催化剂的活性、选择性, 产物分子量、热力学性能等与制备过程密切相关.2002 年, 孟跃中课题组研究了制备方法对合成ZnGA 催化剂的影响.结果表明, 超声处理相比球磨法, 在更大程度上降低了ZnGA[30-31]催化剂的粒径, 增大了比表面积.结晶度相同时, 较大的比表面积可以提供更高的催化活性,产物分子量也相对更高.作者推测戊二酸锌的结晶度和结晶完整性分别是活化CO2的先决条件和提高CO2/PO 共聚活性的关键因素.使用机械搅拌和超声处理后, 催化活性可达160.4 gpoly/gcat, 产物PPC 具有完整的交替结构,Mn为26.3 kDa.孟跃中课题组[31]使用超声法合成的ZnGA, 显著降低了催化剂粒径, 活性达160.4 gpoly/gcat.当催化剂粒径减小后更容易发生颗粒团聚等问题, 催化剂活性仍受到很大的限制[29].
同期, Chisolm 课题组[22]研究发现, ZnGA 催化PO 均聚和CO2/PO 共聚反应中, 共聚物由OH 基团封端, 表明Zn-OH 基团可能是ZnGA 体系中的起始活性物种.在60 ℃、不通CO2情况下, ZnGA 可催化PO 生成规则的首尾相连(HT)结构的聚醚(PPO);在60 ℃, CO2压力(pCO2)为5 MPa 时, ZnGA 可催化CO2/PO 共聚生成PPC, 产率大于97%.在生成的PPC 中, HT 结构约占70%, 首首相连(HH)和尾尾相连(TT)的结构约占30%(图5(a)).作者提出了PO 开环的反应机理:邻近的醇盐基团从背面进攻与Zn 结合的PO 分子, 该醇盐基团与锌中心结合引发了环氧丙烷均聚反应(图5(b)).
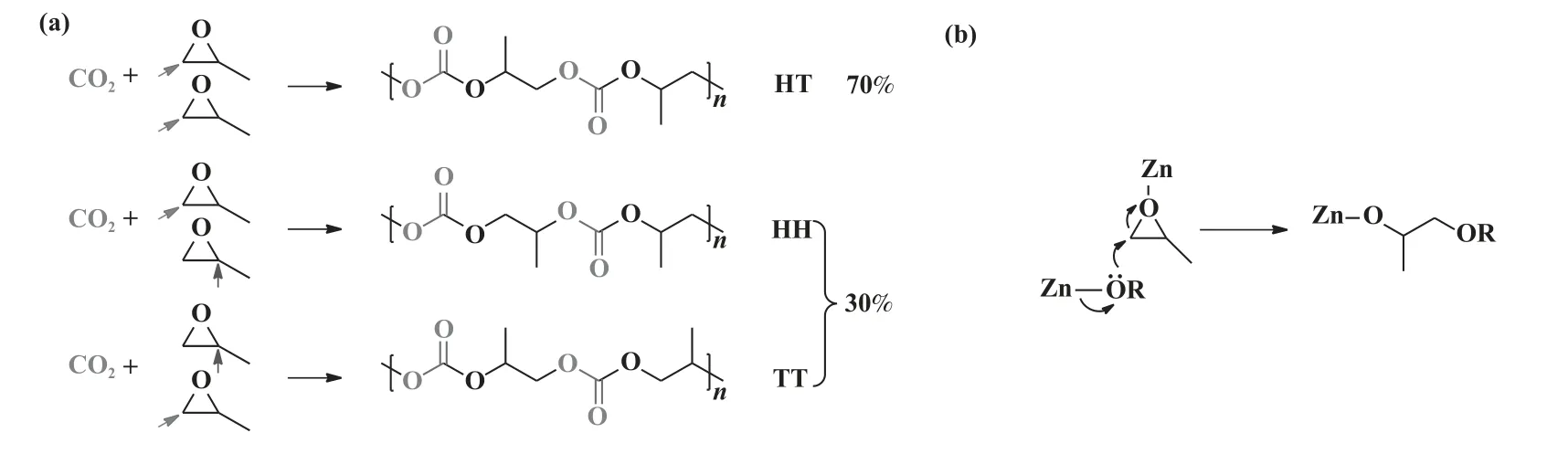
图5 (a) 聚碳酸酯单元的区域序列; (b) 从背面进攻PO 开环反应机理[22]Fig.5 (a) Regiochemistry of polycarbonates; (b) Ring-opening of PO by a back-side attack mechanism[22]
2010 年, Dehghani 课题组[32]研究了超临界CO2对制备ZnGA 催化剂的影响.结果表明, 超临界CO2对催化剂颗粒大小的影响可以忽略不计, 但戊二酸锌的结晶度提高12%, 催化活性提升约10%.Dehghani 课题组[32]开发了一种合成ZnGA 的超临界流体工艺, 催化剂活性从67.4 gpoly/gcat提高到74.2 gpoly/gcat.在超临界CO2中合成ZnGA 的产率超过85%, 与以甲苯为溶剂合成的ZnGA 体系相当.
2017 年, Lee 课题组[33]考察了催化剂制备温度和反应时间对ZnGA 催化剂性能和产物PPC 性能的影响.在70 ℃下, 反应9 h 制备的ZnGA 结晶度达到42.5%, 产物PPC 分子量为99.1 kDa, 分子量分布为1.3.GA 转化率决定了与锌金属中心相连的配体的数量及催化剂活性中心的数量.王嘉骏课题组[34]研究了压力对ZnGA 催化CO2/PO 共聚反应的影响.在低压(0.5~2.6 MPa)下, 随着CO2浓度增加, 共聚速率随着压力的增加而增加.高压(3.5~5.2 MPa)有利于提高PPC 的选择性和聚合物链交替结构的形成.但高压导致催化剂/环氧化物稀释,降低了反应速率.最佳压力范围为2.6~3.5 MPa.2017 年, Salmi 课题组[35]以乙酸锌和戊二酸为原料,在200~250 ℃下研究了原子层沉积法沉积戊二酸锌薄膜.表征结果显示, 在200 ℃下沉积的薄膜呈结晶状态, 其晶体结构与戊二酸锌相匹配.该薄膜可用于催化CO2/PO 共聚反应.
2.4 ZnGA 的改性
ZnGA 具有制备过程简单、选择性高、产物分子量高等优点, 但活性低依旧是ZnGA 工业化所面临的主要问题.Rieger 课题组[29]基于前人的经验总结了ZnGA 改性的方法, 主要包括: (1) 改变搅拌过程; (2) 后修饰; (3) 使用具有高比表面积的添加剂;(4) 调控晶体生长.
2019 年, Yoon 课题组[36]用氯化铁(FeCl3)和氯化锌(ZnCl2)改性的ZnGA 催化CO2/PO 共聚, 活性分别从72.4 提高到90.9 和100.1 gpoly/gcat, 分别提高了25%和38%.随后, Yoon 课题组[37]采用甲醇盐酸溶液蚀刻ZnGA 表面, 形成酸刻蚀的纳米ZnGA催化剂, 活性从72.4 提高到132.1 gpoly/gcat, 提高83%.2015 年, 高利军等[38]采用二氧化硅(SiO2)负载的ZnGA 催化剂, 活性从65.1 提高到131.3 gpoly/gcat.SiO2负载后ZnGA 的比表面积增加, 晶体质量提高, 但整体结晶度下降.为了提高催化剂活性, 研究者将ZnGA 催化剂负载到比表面积较大的材料上, 如蒙脱土[39], MCM-41 分子筛[40], 全氟化合物[41];或者进行酸改性[37], SO2改性[42]等, 这些改性方法在不同程度上提高了ZnGA 的活性(表1).
一般而言, ZnGA 催化剂的表面积越大, 结晶度越高, 则催化活性越高.2005 年, Kim 课题组[43]在合成ZnGA 过程中采用两亲性嵌段共聚物作为有机模板, 以减少颗粒团聚, 提高催化活性.从产物中分离出的ZnGA 催化剂活性低于新合成的催化剂.
2022 年Jang 课题组[44]利用H3[Co(CN)6]活化2D 形态的ZnGA (2D-ZnGA), 催化剂活性提高到855.0 gpoly/gcat, 产物中碳酸酯单元含量为61%, PPC选择性为82.0%,Mn为72.6 kDa,Tg为20.0 ℃.这是目前所报道的活性最高的ZnGA 体系.氰化物的加入对于ZnGA 活性的提升具有积极作用.
将ZnGA 与其他催化剂复合可以有效提高ZnGA 的催化活性, 改善产物性能.2016 年, 刘柏平课题组[45]将ZnGA 与高活性DMC 催化剂按摩尔比10∶1 复合, 催化活性从134.2 增加到508.0 gpoly/gcat.共聚反应产生几乎交替的共聚物, 其碳酸酯单元含量为97.7%,Mn为200.0 kDa, 聚合物多分散指数(PDI)为1.9.共聚反应表现出高活性、高选择性、产物高分子量等特点, 表明ZnGA 和DMC催化CO2/PO 共聚过程中表现出一定的协同作用.
由表1 可见, ZnGA 催化剂通过改性及复配, 催化活性有了一定幅度的提升, 但催化剂活性、 产物分子量和产物性能方面距离工业化应用仍然存在一定的距离[29].尽管如此, 由于ZnGA 催化CO2/PO 共聚反应具有产物PPC 分子量高, 副产物CPC 含量低等特点, 依旧是生产CO2基聚碳酸酯最具工业化潜力的催化剂之一.
2.5 二元共聚反应机理
2011 年, Klaus 等[29]提出了ZnGA 催化CO2/PO共聚反应机理(图6): 该机理涉及戊二酸锌表面上两个活性位点的相互作用, 共聚反应由PO 和CO2交替插入进行, 包括碳酸盐对已配位的PO 亲核进攻和CO2插入锌—醇盐键.
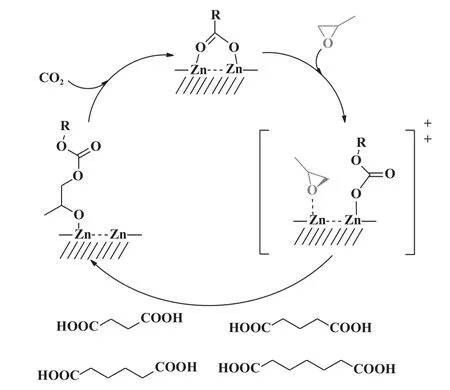
图6 戊二酸锌表面CO2/PO 共聚的双金属反应途径[29]Fig.6 Bimetallic reaction pathway for CO2/PO copolymerization on zinc dicarboxylate surface[29]
通过向反应中加入醇或水等链转移剂(CTA)可使聚合物链发生终止反应, 增长中的链也可以通过链转移与催化剂的活性中心分离.在CO2/环氧化物反应共聚中加入链转移剂可在聚合物末端引入官能团, 如羟基等.2019 年, Darensbourg[46]详细研究了水作为链转移剂在CO2/环氧化物共聚反应中的作用和功能(图7).

图7 PO 和CO2 在水存在下的永生共聚反应[46]Fig.7 Immortal copolymerization of PO and CO2 in the presence of water[46]
CO2/环氧化物共聚通常通过一个或两个金属中心的配位—插入机理进行[47-48], 主要包括3 个基本步骤(图8): (1)链引发, (2) 链增长, (3) 链终止.一分子环氧化物或CO2在金属活性中心上配位插入形成金属醇盐或金属碳酸盐中间体, 引发基团连接到增长链的末端.另一分子CO2或环氧化物插入金属—氧键继续进行链增长反应.两种单体交替插入形成碳酸酯键.然而, 在反应过程中可能发生两种副反应[29,49]: (1) 发生回咬反应(backbiting reaction)形成环状碳酸酯CPC, (2) 环氧化物分子连续插入生成醚键.两个CO2分子的连续插入因反应能垒较高而不能发生[49].
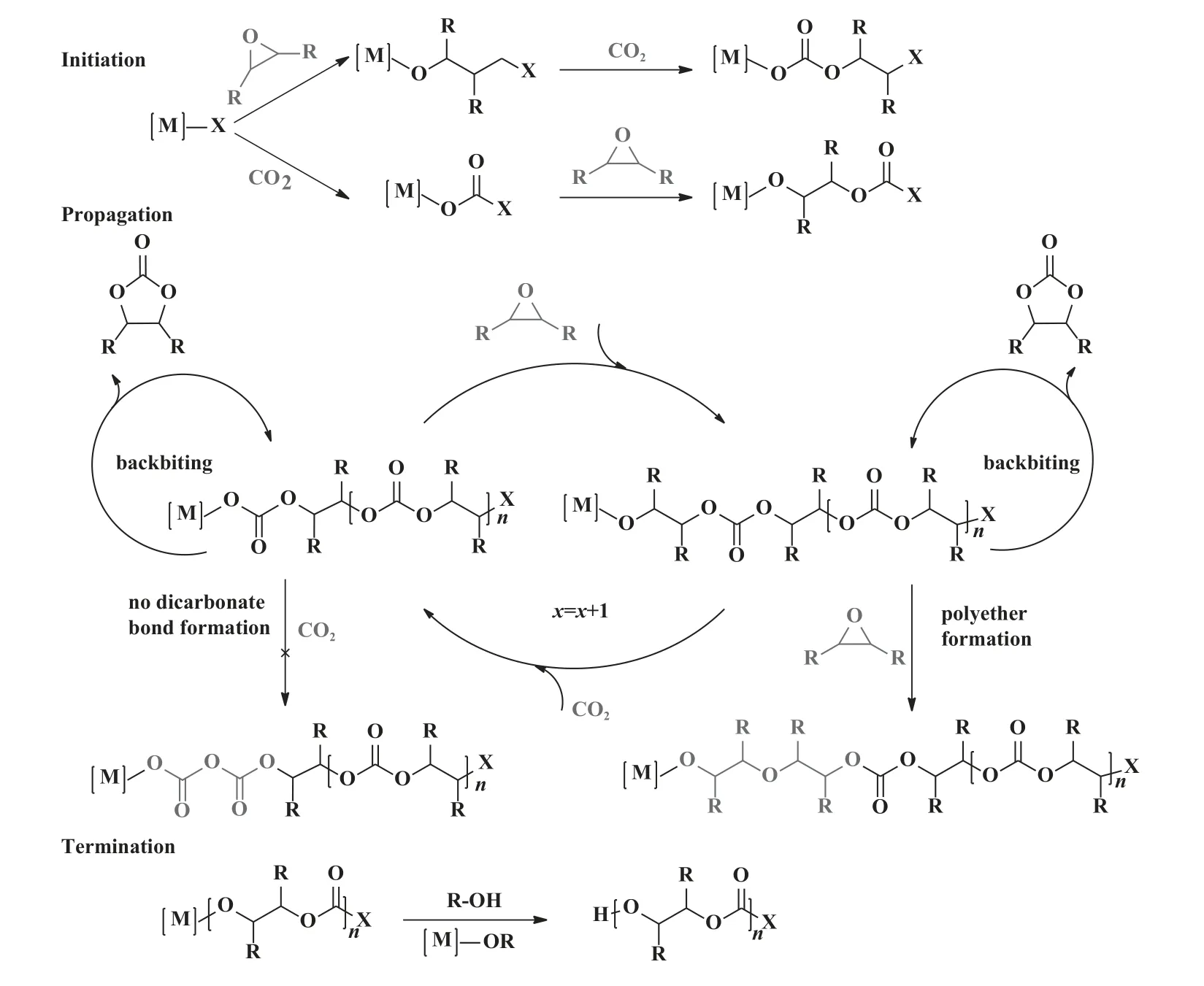
图8 CO2/环氧化物开环共聚反应机理[47-48]Fig.8 General mechanism of the ring opening copolymerization of epoxides and CO2[47-48]
3 ZnGA 催化CO2/PO/第三单体共聚
CO2/PO 二元共聚产物的玻璃化转变温度(<40.0 ℃)相对较低, 在共聚中引入第三单体可以有效提高产物的热力学性能和机械性能.马来酸酐是CO2/PO/第三单体三元共聚中常见的第三单体.除此之外, 还有邻苯二甲酸酐(PA)、 衣康酸酐(IAn)等, 结构式如图9 所示.通过改变反应条件和单体之间的摩尔比, 可以调控三元共聚产物的玻璃化转变温度.其次, PPC 链反咬形成的副产物CPC,其沸点高达242.0 ℃, 难以通过热处理去除, 第三单体的引入可有效降低聚合产物中CPC 的含量.
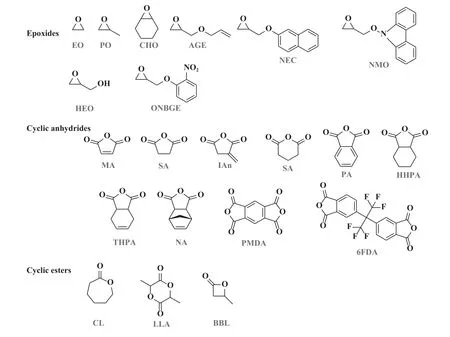
图9 常见的环氧化物、 环状酸酐和环酯的结构Fig.9 Structures of commonly applied epoxides, cyclic anhydrides and cyclic esters
3.1 CO2/PO/环状酸酐三元共聚
2008 年, 孟跃中课题组[50]利用全氟化合物负载的ZnGA 催化剂实现了CO2/PO/马来酸酐三元共聚, 在60 ℃、pCO2为5.2 MPa 的条件下反应40 h,得到共聚产物聚马来酸碳酸丙烯酯(PPCMA).在环氧丙烷与马来酸酐摩尔比为30∶1 时, 产物PPCMA301 的Mn为67.1 kDa,PDI为3.28, 催化剂活性为72.5 gpoly/gcat.随着马来酸酐加入量的增加,PPCMA 的热稳定性显著提高.随后, Song 等[51]使用过氧化二异丙苯(DCP)作为交联剂, 使含有少量马来酸酐的PPCMA 发生交联.与未交联的PPCMA 相比, 热性能和机械性能有显著提高.含有DCP 质量分数为0.6%的PPCMA 的5%失重温度(Td,-5%)和最大失重温度(Td,max)分别为261.0 和300.0 ℃.含有DCP 质量分数为0.8%的PPCMA的Tg为42.9 ℃, 拉伸强度为45.59 MPa.
2014 年, 孟跃中课题组[52]报道了ZnGA 催化CO2/PO/邻苯二甲酸酐三元共聚, 在75 ℃,pCO2为5 MPa, 马来酸酐与环氧丙烷摩尔比为1∶8 条件下,反应15 h 得到CO2/PO/邻苯二甲酸酐共聚产物,Mn为210.0 kDa,PDI为2.8, 催化剂活性为52.8 gpoly/gcat, 分子量分布较宽, 且聚合产物中存在一定量的醚键(3.3%~5.1%(摩尔分数)).由于邻苯二甲酸酐插入Zn-OH 的速率很慢, 导致CO2的反应速率(rCO2=5.94)显著高于邻苯二甲酸酐, 产物为无规则结构.
宋鹏飞课题组[53]报道了ZnGA 催化CO2/PO/衣康酸酐三元共聚, 在70 ℃,pCO2为5.2 MPa, 衣康酸酐与环氧丙烷摩尔比为1∶20 条件下, 反应40 h获得三元共聚产物PPCIAn,Mn为101.0 kDa,PDI为2.8, 催化剂活性为42.3 gpoly/gcat.Td,-5%为257.8 ℃,Td,max为286.1 ℃.随着衣康酸酐与环氧丙烷摩尔比增加, 催化活性提高大约10.0 gpoly/gcat.
2013 年, 丰九英课题组[54]报道了CO2/PO/均苯四甲酸二酐(PMDA)三元共聚, 在70 ℃,pCO2为5.4 MPa 条件下, 反应36 h 得到共聚产物, 均苯四甲酸二酐相对PO 的添加量不超过4%(摩尔分数)的产物表现出优异的热性能和机械性能,Td,-5%大于270.0 ℃,Td,max大于288.0 ℃, 拉伸强度为26~45 MPa.催化活性最高为47.3 gpoly/gcat, 重均分子量高达180.0~860.0 kDa.2019 年, Gao 等[55]成功地将马来酸酐均聚物(MAO)引入PPC 主链.MAO 的质量比从0 增加到3.75%, 产率从26.0 提高到67.0 gpoly/gcat,Td,-5%和Td,max分别从215.0 和256.0 ℃提高到287.7 和302.5 ℃.但Tg最大值仅为36.2 ℃,马来酸酐均聚物的引入并不能明显改善聚合物的力学性能.
2015 年, Luinstra 课题组[56]以ZnGA 为催化剂,将琥珀酸酐(SA)、 马来酸酐、 邻苯二甲酸酐、 戊二酸酐、 5-降冰片烯-2,3-二羧酸酐、 六氢邻苯二甲酸酐(HHPA)、 四氢邻苯二甲酸酐(THPA)引入CO2/PO 二元共聚体系, 在60 ℃,pCO2为3 MPa 条件下反应4 h.相比CO2/PO 二元共聚反应, 第三单体的加入将产物的重均分子量从210.0 提高到520.0 kDa, 同时降低了副产物CPC 的含量.酸酐浓度在0.3%(摩尔分数)时, 5-降冰片烯-2,3-二羧酸酐的加入使CPC 从4.5%降低至约1%.第三单体的加入减缓了反咬反应的速率, 降低了CPC 的含量(图10).
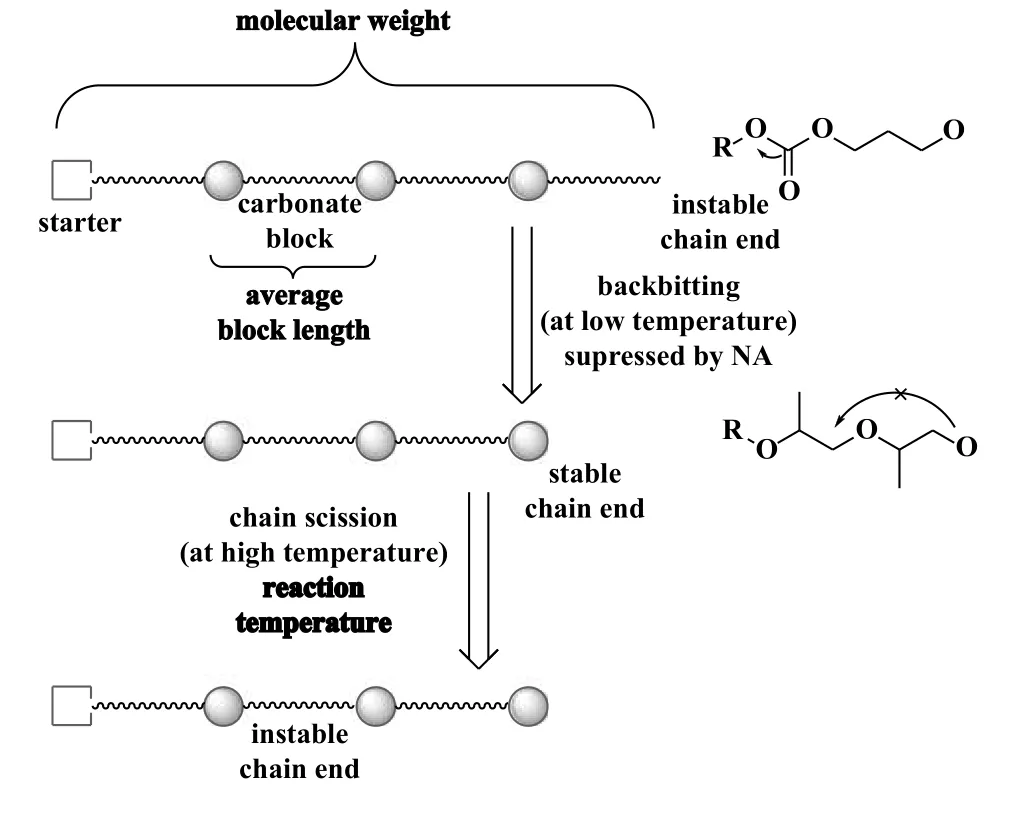
图10 反咬反应生成CPC 和PPC 链断裂的影响[56]Fig.10 CPC formation by backbiting and role of chain scission in PPC[56]
2021 年, 王文珍课题组[57]以ZnGA 催化CO2、PO 和4,4′(六氟异丙基)二苯二甲酸酐(6FDA)合成聚碳酸酯, 催化活性为104.0 gpoly/gcat,Mn为212.0 kDa,Td,-5%和Td,max分别为320.0 和350.0 ℃,Tg高达52.0 ℃.刚性基团苯环的引入和交联网络提高了产物的稳定性和热性能.
3.2 CO2/PO/环酯三元共聚
2003 年, Ree 课题组[58]实现了ZnGA 催化CO2/PO/己内酯(CL)三元共聚(图11).在T为60 ℃,pCO2为2.7 MPa 条件下反应40 h, 合成的半结晶梯度共聚物中没有检测到副产物CPC, 但存在一定比例的己内酯均聚产物, 且随着己内酯与环氧丙烷摩尔比从3∶7 增加到8∶2, 己内酯均聚物的含量从13.7%增加到64.5%, 催化活性从47.7 降低到20.2 gpoly/gcat, 产物分子量呈先上升后下降的趋势.在己内酯与环氧丙烷摩尔比为5∶5 时,Mw最高为369.0 kDa.2016 年, 孟跃中课题组[59]采用全氟化合物负载的ZnGA 催化CO2/PO/CL 三元共聚.随着己内酯与环氧丙烷摩尔比从1∶9 增加到3∶7, 催化活性从50.0 增加到102.0 gpoly/gcat, 产物Mw仅为87.0 kDa,PDI为1.7.该反应时间在27 h 之前, 己内酯插入主链速率大于CO2, 倾向于生成PCL; 27 h之后, 随着CL 不断消耗, 环氧丙烷与己内酯摩尔比不断升高, 倾向于生成PPC.
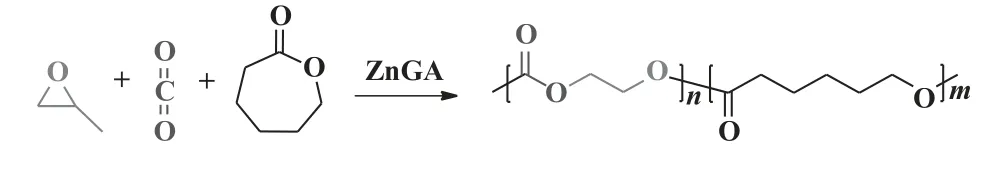
图11 ZnGA 催化CO2/PO 和CL 三元共聚[58]Fig.11 Terpolymerization of CO2/PO and CL catalyzed by ZnGA[58]
2017 年, 宋鹏飞课题组[60]报道了ZnGA 催化CO2/PO/L-丙交酯(LLA)三元共聚.随着L-丙交酯与环氧丙烷摩尔比从1∶50 增加到1∶10, 催化活性从43.3 提高到48.4 gpoly/gcat.Mn从76.0 提高到120.0 kDa,Tg从39.1 提高到40.9 ℃,Td,-5%和Td,max分别从253.5 和271.4 ℃提高到260.1 和281.4 ℃.三元共聚产物的热稳定性和机械性能较PPC 有显著提高, 但L-丙交酯插入率小于6%.
2021 年, 庞烜课题组[61]设计了一种包含Salen CoⅢ、 ZnGA 和双三苯基膦氯化铵(PPNCl)的三元复合催化剂催化CO2/PO/LLA 三元共聚, 合成了含有聚乳酸和CO2基聚碳酸酯的多嵌段共聚物.产物中聚乳酸含量为69%, 共聚产物Mn高达698.0 kDa.这是目前报道的聚乳酸和PPC 的多嵌段共聚物的最高值.嵌段共聚产物可能存在成分漂移现象,导致存在多个Tg值.相比于单独使用ZnGA 或者己二酸锌[62-63]产生的交替共聚物, 存在性能差异, 但其共聚产物具有较高的分子量.
2018 年, Yoon 课题组[64]首次使用纳米ZnGA催化剂, 并成功应用于CO2/PO/丁内酯(BBL)三元共聚.调节丁内酯与环氧丙烷的进料比从1∶9 提高到1∶3, 催化活性从24.0 降低到9.5 gpoly/gcat, 玻璃化转变温度从34 降低到20 ℃, 丁内酯的最大插入率为29%.
3.3 CO2/PO/其它单体三元共聚
除了环状酸酐、 环酯可作为第三单体参与共聚反应之外, 环氧环己烷(CHO)、 环氧乙烷(EO)和缩水甘油醚等也可以作为第三单体进行三元共聚.2011 年, 孟跃中课题组[65]利用ZnGA 催化CO2/PO/CHO 共聚反应, 成功地合成了三元交替共聚产物.催化活性为128.1 gpoly/gcat,Mn为143.0 kDa,PDI为2.3.将少量的环氧环己烷引入PPC 主链,Tg从38.0 提高到43.0 ℃.当环氧乙烷作为第三单体参加三元共聚时[66], 产物的催化活性、 分子量和玻璃化转变温度均出现不同程度的降低.作者还研究了N-(2,3-环氧丙基)咔唑(NEC)[67]和[(2-萘氧基)甲基]环氧乙烷(NMO)[68]作为第三单体的共聚反应,当加入4.3%(摩尔分数)的NEC 作为第三单体时,聚合活性为60.0 gpoly/gcat,Mn为74.0 kDa.加入6.2%的NMO 时, 聚合活性为61.0 gpoly/gcat,Mn为35.0 kDa.
2014 年Luinstra 课题组[69]利用ZnGA 催化CO2/PO/邻硝基苄基缩水甘油醚(ONBGE)的三元共聚, 生成具有功能性侧基的PPC(图12).通过紫外照射完全去除PPC 主链中的邻硝基苄基, 合成了带有羟基基团的功能化PPC, 具有显著的亲水性.
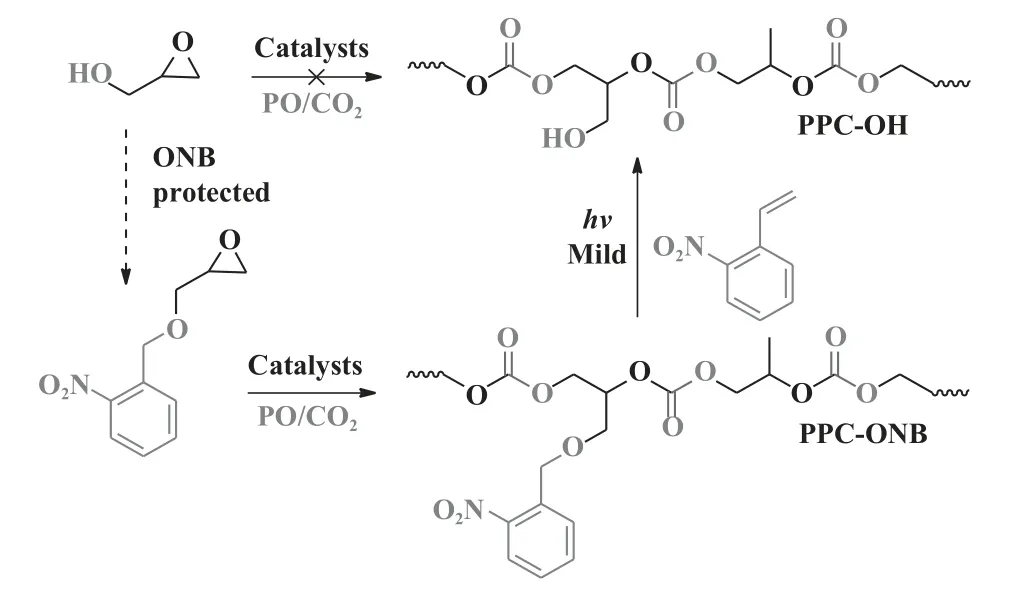
图12 合成羟基功能化聚碳酸酯[69]Fig.12 Synthesis of hydroxyl-functionalized poly(propylene carbonate)[69]
表2 总结了常见的戊二酸锌催化CO2/PO/第3单体的三元共聚反应.总体而言, 加入第3 单体会在不同程度上降低催化剂的活性, 但在不过度损失催化活性的基础上, 添加一定量的第3 单体, 可以达到改善产物性能、 降低副产物含量等目标.虽然研究者进行了大量尝试, 但完美交替的三元共聚物的合成极其困难, 几乎所有的产物都存在不同程度的嵌段共聚.一锅法在一定程度上改进了聚碳酸酯的性能, 拓宽了其应用范围.
3.4 三元共聚反应机理
孟跃中课题组[70]提出了一种关于ZnGA 催化生成聚碳酸酯和聚酯的阴离子配位机理.对于环氧化物、 CO2和环状酸酐一锅共聚, 其机理如图13 所示.依照该反应机理, 环氧化物的C—O 键首先发生断裂, 插入到催化剂的金属活性中心, 形成金属醇盐结构1; CO2和环状酸酐竞争插入1, 分别生成金属碳酸盐2 和金属羧酸盐3; 环氧化物插入2 或3, 重复上述循环.对于环氧化物、 CO2和环酯一锅共聚,环酯开环插入后, 生成了与金属羧酸盐3 类似的金属醇盐中间体.
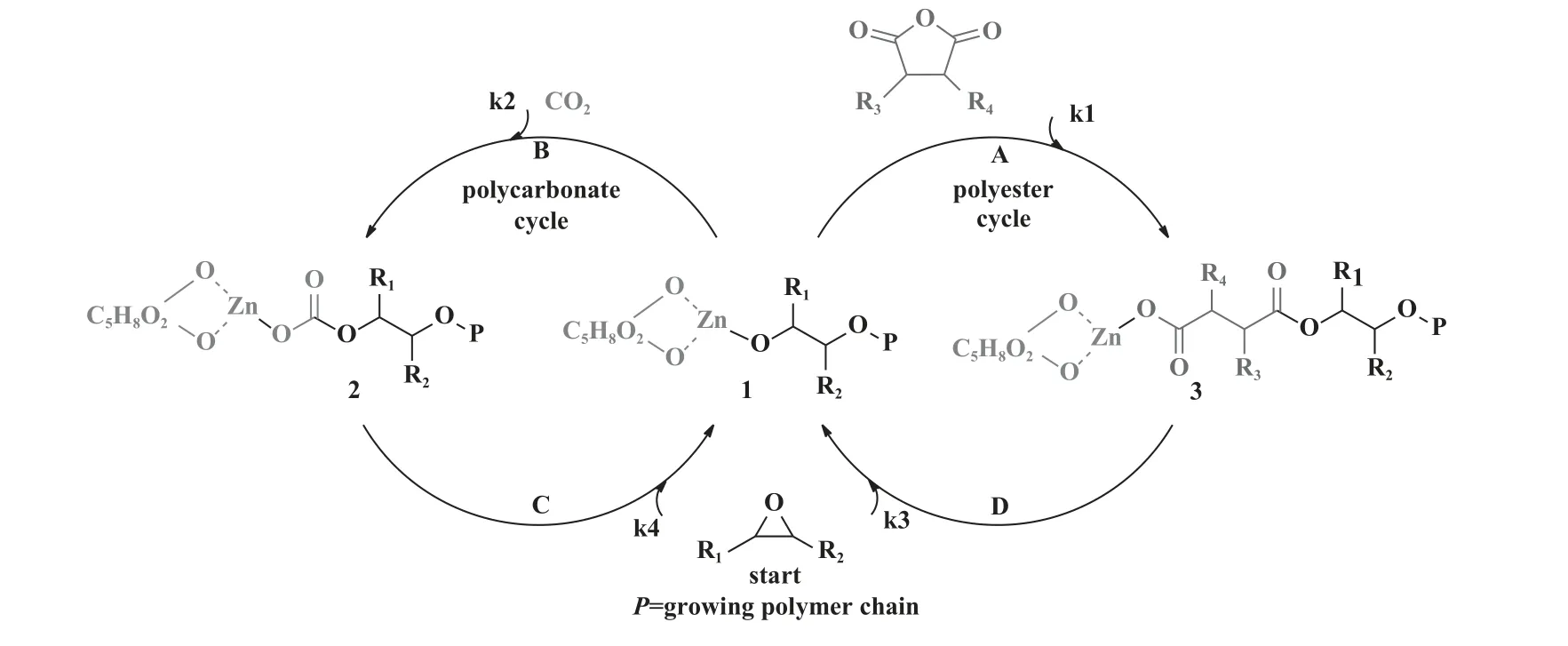
图13 CO2、 环氧化物和环状酸酐共聚反应机理[70]Fig.13 Mechanism of the copolymerization of epoxides, CO2 and cyclic anhydrides via one-step procedure[70]
应注意的是, 只有通过一锅/一步工艺共聚遵循上述机理.k1~k4分别对应A~D 过程的速率常数k, 其大小决定插入速率, 两个循环的速率差异又决定聚合物的微观结构, 其中, 速率决定步骤为环氧化物的插入反应.因此, 动力学分析对于聚合物微观结构的解析至关重要.
2011 年, 孟跃中课题组[65]提出了CO2/PO/CHO共聚反应机理(图14).与PO 和CHO 相比, CO2更容易与催化剂的Zn 金属活性中心配位, 并插入到Zn—O 键中, 生成碳酸盐中间体; 然后PO 和CHO竞争与催化剂的Zn 金属活性中心配位, Zn 活性中心被碳酸盐阴离子攻击, 速率决定步骤为PO 和CHO 的插入反应; 随后CO2、 PO 和CHO 的交替插入形成聚碳酸酯.
4 总结与展望
CO2是稳定无毒、 廉价易得、 来源广泛的C1资源.由CO2和环氧化物形成的聚合物具有热塑性、稳定性高、 透明和环境友好等优点, 在消耗CO2的同时带来一定的经济效益.我们综述了戊二酸锌催化CO2和环氧化物二元共聚和三元共聚, 以及催化机理等方面的研究.
尽管戊二酸锌催化剂取得了显著进步并表现出优良性能, 但仍需进一步提高其催化活性, 深入研究其催化机理, 着力解决工艺过程中易产生环状碳酸酯副产物等问题.另一方面, 在结构和性能关系方面, 仍缺乏CO2基聚碳酸酯的力学、 结晶和降解等性能方面的研究, 而这对聚合物的加工应用至关重要.因此, 需要深入研究CO2/PO 共聚及环状副产物的生成机理, 构建CO2基聚碳酸酯的结构和性能的关系, 在学术界和工业界的共同努力下, 最终实现CO2基聚碳酸酯的绿色合成及低成本工业规模生产.
猜你喜欢
新作文·中学作文教学研究(2022年4期)2022-08-25 02:38:48
太原科技大学学报(2020年3期)2020-06-22 01:27:26
中国塑料(2016年2期)2016-06-15 20:29:59
航天制造技术(2016年6期)2016-05-09 08:32:48
中国塑料(2016年5期)2016-04-16 05:25:39
支点(2015年11期)2015-11-16 10:25:03
云南中医学院学报(2015年1期)2015-07-31 18:10:45
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
无机化学学报(2014年4期)2014-02-28 17:31:23
