
V-Ce/TiO2 脱硝催化剂的SO2 中毒机理研究
2024-01-04 11:56:56郝广源
分子催化 2023年5期
郝广源, 井 宇
(北京太阳宫燃气热电有限公司, 北京 100028)
锅炉系统中使用的化石燃料的燃烧会产生大量的工业燃烧氮氧化物(NO、 NO2), 其排放会直接威胁到生态系统和人类健康.使用NH3的选择性催化还原(NH3-SCR)方法已被广泛认为是去除氮氧化物的最有效和最经济的技术[1], 该技术采用V2O5-WO3/TiO2催化剂, 并在较高的温度区间内(300~400 ℃)进行NOx的选择性催化还原反应.近年来,由于工业窑炉等非电行业锅炉系统的减排需求, 结合其烟道空间限制, 大量的研究聚焦在低温NH3-SCR 脱硝催化剂的研发上, 如Ce 基[2]、Mn 基[3]、Fe基等[4].国内企业如北京方信立华、合肥晨曦、上海瀚昱等环保企业已研发出具有独立知识产权的低温脱硝催化剂, 并已成功应用于钢铁、冶金、制药、水泥等行业[5-7].
然而, 烟气中大量SO2的存在进一步限制了该低温NH3-SCR 脱硝催化剂的应用.大量研究结果表明, 在含SO2体系下, Ce、Mn、Fe 基催化剂低温脱硝活性呈断崖式下跌.Jin 等[8]发现Mn/TiO2催化剂在150 ℃的含SO2氛围下10 h 后活性下降至50%;Ma 等[9]发现Fe-Cu/TiO2催化剂在200 ℃含SO2氛围下50 min 后活性仅剩3%; Zhang 等[10]研究表明570 mg/L SO2存在条件下, 240 ℃脱硝活性从85%下降至20%.以上研究结果表明Ce、Mn、Fe基催化剂在SO2氛围下存在严重的低温失活问题,无法实现稳定的低温脱硝反应.不少研究重新聚焦在V2O5基催化剂的研发上, Li 等[11]研究了VPd/TiO2催化剂中低温抗硫中毒NH3-SCR 性能, 表征结果表明V2O5未被硫酸化, 且DFT 计算表明V2O5与SO2没有化学吸附作用.Xu 等[12]对比研究了V2O5-WO3/TiO2和CeO2-WO3/TiO2催化剂的抗SO2中毒性能, 结果发现V2O5-WO3/TiO2催化剂活性下降主要是由于硫酸铵盐的沉积, 且催化剂经热再生后仍能恢复脱硝活性, 而CeO2-WO3/TiO2催化剂由于大量金属硫酸盐的生成造成永久失活.Kwon 等[13]对V-Sb/TiO2和V-Sb-Ce/TiO2催化剂进行了中毒对比研究, 结果发现中毒后V-Sb/TiO2沉积的硫物种以硫酸铵盐为主, 而中毒后V-Sb-Ce/TiO2产生了大量硫酸铈.Maqbool 等[14]对硫酸化后Sb-Ce-V2O5/TiO2进行了活性测试, 结果表明预硫化后催化剂表面仅发现了Ce2(SO4)3和Ce(SO4)2物种, 通过酸性的增加提升了催化剂脱硝活性.
以上结果表明, 催化剂活性组分V2O5具有较好的抗SO2中毒能力, 我们以V-Ce/TiO2催化剂为目标, 考察其在SO2氛围下、不同反应温度下的脱硝活性演变, 结合XPS、TG 等表相体相定量手段,定量地获得金属硫酸盐、硫酸铵盐在不同反应温度下的沉积量, 进一步结合原位红外分析手段辨析了V-Ce/TiO2催化剂在不同温度下的中毒机理, 为V基催化剂在含硫氛围低温脱硝的工业化应用提供理论指导.
1 实验部分
1.1 催化剂制备
V-Ce/TiO2(V/Ti 摩尔比为0.1)催化剂采用溶胶凝胶法制备, 具体流程如下: 取5 g TiO2粉末(阿拉丁, 99%)分散于50 mL 去离子水中, 置于超声分散器中分散30 min, 加入一定量的偏钒酸氨(阿拉丁,99%)和硝酸铈粉末(阿拉丁, 99%), 并加入4 g 柠檬酸(阿拉丁, 99.5%), 置于磁力搅拌器中常温搅拌2 h, 70 ℃下恒温搅拌直至水分完全蒸发, 溶胶样品放于85 ℃的烘箱中干燥24 h, 最后将粉末样品放入马弗炉中, 600 ℃煅烧6 h, 并研磨至0.450~0.180 mm 备用.
Ce(SO4)2/TiO2(V/Ti 摩尔比为0.1)催化剂采用浸渍法制备, 具体流程如下: 取5 g TiO2粉末(阿拉丁, 99%)分散于50 mL 去离子水中, 置于超声分散器中分散30 min, 加入硫酸铈粉末(阿拉丁, 99%),70 ℃下置于磁力搅拌器搅拌直至水分完全蒸发, 样品干燥及煅烧过程与V-Ce/TiO2催化剂相同.
1.2 催化剂活性
催化剂性能测试在固定床反应器中进行, 石英反应管, 内径17 mm, NO、NH3、SO2、O2、N25 路气体均采用质量流量计精确控制流量, 并使用内径35 mm 的圆柱形预混器充分混合, 其余管路均为3 mm 不锈钢气管.测试流程如下: 0.2 g 催化剂置于反应管石英棉床层, 随后通入N2并以10 ℃/min 升温至目标反应温度, 随后通入反应气体, 浓度为1 340 mg/L NO、 760 mg/L NH3、570 mg/L SO2(抗硫、 抗硫抗水测试时通入)、10% H2O(抗硫抗水测试时通入)、5%O2及高纯N2作为平衡气, 总流量为700 mL/min, 空速为42 000 h-1.尾部烟气通入经Testo350 烟气分析仪、 Medi-Gas G200 对出口NOx、N2O 进行连续分析, NOx的转化率(η)和N2O 选择性(S)计算公式如下:
其中[NOx]out、 [NOx]in分别表示出口的NO 和NO2、进口的NO 和NO2.
1.3 催化剂表征
XRD 测试采用Philips X pert Pro 衍射仪, 0.08 g研磨后的催化剂粉末置于石英板上并压平, 后插入插槽, 以5 (°)/min 速率扫描样品; BET 测试采用VSorb 2800P 物理吸附分析仪, 0.1 g 样品仅250 ℃,6 h 脱气处理后进行吸脱附分析, 获得比表面积与孔隙结构等信息; XPS 测试采用PHI Quantera II 仪器,0.02 g 样品经抽真空后进行测试, 结合能以污染碳284.6 eV 进行矫正; TG-DTG 测试采用HCT-2 热重分析仪, 0.03 g 样品置于小坩埚内, N2保护下升温同时记录样品质量变化; NH3-TPD 与H2-TPR 测试均采用彼奥德化学吸附分析仪, 50 mg 样品经250 ℃预处理1 h 后, 降温至常温下通入30 mL/min 的10% NH3/He, 充分吸附1 h 后切换成He 气常温吹扫0.5 h, 随后以10 ℃/min 升温至900 ℃, 脱附的NH3经过TCD 热导传感器记录; H2-TPR 测试过程如下: 首先催化剂在250 ℃下预处理1 h, 随后降温常温通入20% H2/N2走平TCD 基线, 随后以10 ℃/min 升温至600 ℃, 消耗的H2经TCD 热导检测器记录; 原位漫反射傅里叶变换红外光谱(in-situ DRIFTS)测试采用 Nicolet 6700 光谱仪结合Harrick IR 池和一个MCT 检测器, E-R 反应路径的DRIFTS 测试流程如下: 催化剂样品放入原位反应池中, 在20 mL/min 的N2气氛下吹扫1 h, 随后降温至180 ℃, 通入76 mg/L 的NH3, 吸附40 min 保证吸附饱和, 随后通入N2吹扫10 min, 记录谱图, 最后通入134 mg/L 的NO 与5% O2, 每隔5 min 记录谱图.L-H 反应路径的DRIFTS 测试流程如下: 催化剂样品放入原位反应池中, 在20 mL/min 的N2气氛下吹扫1 h, 随后降温至180 ℃, 通入134 mg/L 的NO 与5% O2, 吸附40 min 保证吸附饱和, 随后通入N2吹扫10 min, 记录谱图, 最后通入76 mg/L 的NH3, 每隔2 min 记录谱图.
2 结果与讨论
2.1 催化剂活性评价
不同Ce 掺杂比例的V-Ce/TiO2催化剂活性及抗硫性能如图1 所示, 从图1(a)中可以看出, 随着Ce/Ti 摩尔比从0 增加至0.2, 催化剂低温活性呈现先增大后减小的变化规律, 这可能是由于过高的Ce 负载量不利于活性金属氧化物的分散, 导致活性不增反降.当Ce/Ti 摩尔比为0.1 时, V-Ce/TiO2催化剂具有最高的低温活性, 180 ℃下脱硝效率可达77.4%, 210 ℃及更高反应温度下, 脱硝活性可达100%.N2O 选择性如图1(b)所示, 从图中可以看出随温度升高, N2O 选择性不断增加, 且随着Ce 负载量的增加, 催化剂N2O 选择性增加.为了探究VCe(0.1)/TiO2催化剂在不同温度下的抗硫性能, 含硫氛围下8 h 内的脱硝活性演变如图1(c)所示, 180 ℃下, 随着SO2通入1 h 后, 脱硝活性从初始的77.4%下降至67.7%, 随着反应时间的进一步增加, 脱硝活性不断缓慢下降, 8 h 后活性下降至50.0%.当反应温度为240 和300 ℃时, V-Ce(0.1)/TiO2催化剂的脱硝活性在8 h 内均稳定在100%.10% H2O 与SO2共存气氛条件下, 不同反应温度的脱硝活性演变如图1(d)所示, 从图中可以看出180 ℃、 8 h 后脱硝活性降低至35.1%, 240 ℃、 8 h 后脱硝活性降低至87.1%, 300 ℃、 8 h 后脱硝活性降低至94.2%.以上结果表明V-Ce(0.1)/TiO2催化剂的抗硫性能极度依赖于反应温度.
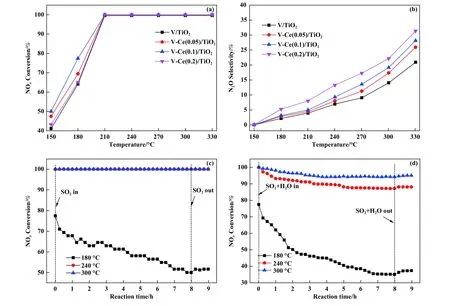
图1 V-Ce(0, 0.05, 0.1, 0.2)/TiO2 催化剂的NOx 转化率和N2O 选择性((a), (b)); 不同温度下SO2 氛围下NOx 转化率随时间演变(c); 不同温度下SO2+H2O 共存下NOx 转化率随时间演变(d)Fig.1 NOx Conversion and N2O selectivity over fresh V-Ce (0, 0.05, 0.1, 0.2)/TiO2 catalysts ((a), (b)); evolution of NH3-SCR activity with time on stream in the presence of SO2 (c) and evolution of NH3-SCR activity with time on stream in the presence of SO2 and H2O (d)
2.2 中毒前后物化特性分析
2.2.1 晶相与结构参数分析
为了探究不同反应温度后催化剂的晶相变化,XRD 图谱如图2(a)所示, 从图中可以看出反应前后催化剂无明显晶相变化, 衍射峰归属于锐钛型TiO2晶体(PDF#21-1 272)、CeVO4晶体(PDF#12-0757).为了进一步探究可能存在的硫酸铵盐、金属硫酸盐等对催化剂结构性质的影响, 孔径分布及比表面等结构参数如图2(b)、 表1 所示, 从图中可以看出中毒前后催化剂的孔径分布无明显变化, 最可几孔径位于23.78 nm.结构参数如表1 所示, 从表中可以看出新鲜V-Ce(0.1)/TiO2催化剂的比表面积为47.1 m2/g, 孔容为0.21 cm3/g, 平均孔径为18.6 nm,经过不同反应温度下的中毒实验后, 催化剂比表面积、孔容、平均孔径均有所下降.以上结果表明180 ℃下, SO2与NO 的竞争吸附不是活性下降的主要原因, 催化剂结构性质的改变导致了催化剂明显的低温失活现象.
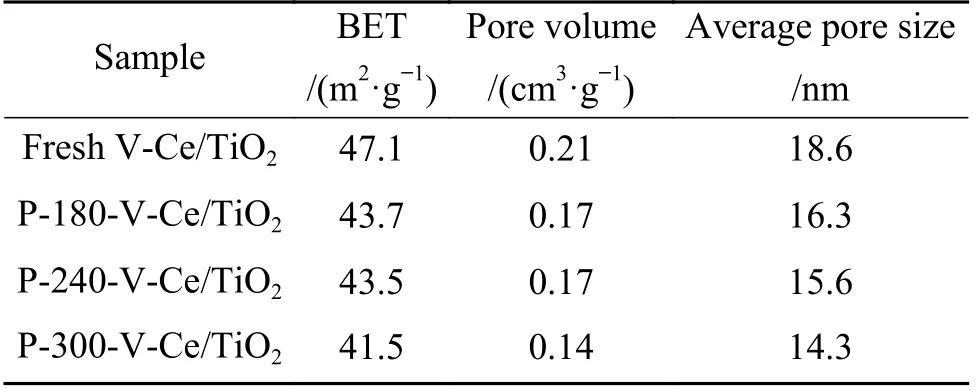
表1 新鲜以及中毒后V-Ce(0.1)/TiO2 催化剂的结构参数Table 1 Structural parameters of fresh and poisoned V-Ce(0.1)/TiO2 catalysts
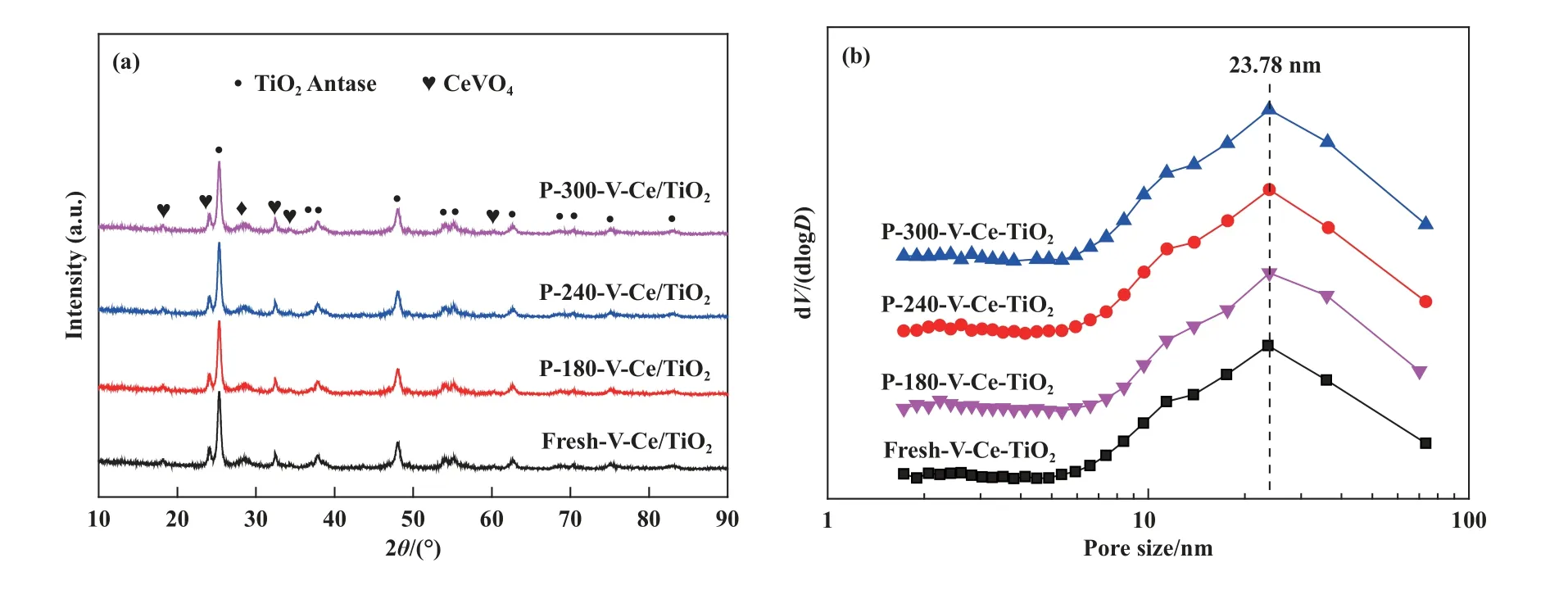
图2 V-Ce(0.1)/TiO2 催化剂在不同温度中毒后的XRD 谱图(a)及孔径分布图(b)Fig.2 XRD Pattern of V-Ce(0.1)/TiO2 catalysts after SCR reaction at different temperatures in SO2-containing atmosphere (a) and pore size distribution (b)
2.2.2 体相硫组分TG-DTG 分析
为了进一步获得催化剂体相中含硫组分的定量结果, TG-DTG 测试被用于判断含硫组分及其所占比重, TG-DTG 如图3 所示, 从图3(a)中可以看出180 ℃中毒后催化剂表现出3 个DTG 峰, 分别位于410~555 ℃、555~760 ℃、 760~930 ℃ 3 个区间温度内, 其中410~555 ℃的失重峰可以归结于NH4HSO4的分解[15].为了进一步探究555~760 ℃、760~930 ℃失重峰归属情况, 我们制备了Ce/TiO2、V/TiO2催化剂并进行了SO2+O2氛围下的硫酸化实验, 随后进行TG-DTG 测试, 结果如图3(b)所示, 硫酸化后的Ce/TiO2催化剂失重温度区间为710~840 ℃, 硫酸化后的V/TiO2催化剂失重温度区间为630~700 ℃.这里推测图3(a)、(c)、(d)中的555~760℃的失重峰可能归属于硫酸氧钒[16], 760~930 ℃的失重峰可能归属于硫酸铈的分解[17], 以上3 个含硫组分的失重占比分别为0.48%、2.38%、 0.83%.随着反应温度升高至240 ℃, 硫酸氢铵、 硫酸氧钒、硫酸铈的失重占比分别为0.27%、2.81%、0.89%, 随着反应温度进一步升高至300 ℃, 硫酸氢铵的失重峰消失, 硫酸氧钒、硫酸铈占比分别增加至3.53%、0.92%.以上结果表明随着反应温度的增加, 催化剂上沉积的硫酸氢铵逐渐减少, 金属硫酸盐逐渐增多.
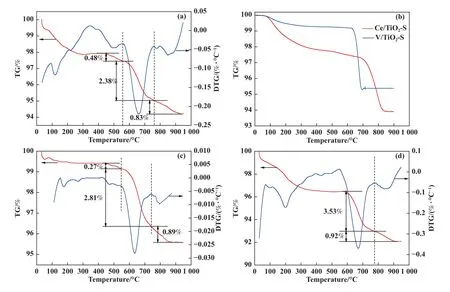
图3 不同反应温度下中毒后的V-Ce(0.1)/TiO2 催化剂的TG-DTG 图谱(a) 180 ℃反应后; (b) 硫酸化后V/TiO2 及Ce/TiO2; (c) 240 ℃反应后; (d) 300 ℃反应后Fig.3 TG-DTG Pattern of V-Ce(0.1)/TiO2 catalysts after SCR reaction at different temperatures in SO2-containing atmosphere(a) at 180 ℃; (b) Sulfated V/TiO2 and Ce/TiO2; (c) at 240 ℃; (d) at 300 ℃
2.2.3 沉积的硫酸氢铵分析
催化剂粉末中沉积的硫酸氢铵含量进一步通过纳氏试剂-分光光度计法[18]进行测试, 结果如图4(a)、(b)、(c)所示.180 ℃下随着反应时间的增加, 铵盐不断在催化剂表面沉积, 8 h 后NH4+浓度高达0.075 mmol/g.240 ℃下浓度明显降低, 8 h 后浓度仅为0.032 mmol/g.随着反应温度进一步上升至300 ℃, 8 h 中毒反应后浓度仅剩0.001 7 mmol/g.以上结果进一步表明低温下硫酸铵盐在催化剂表面大量沉积, 随着温度升高铵盐的沉积量明显降低.
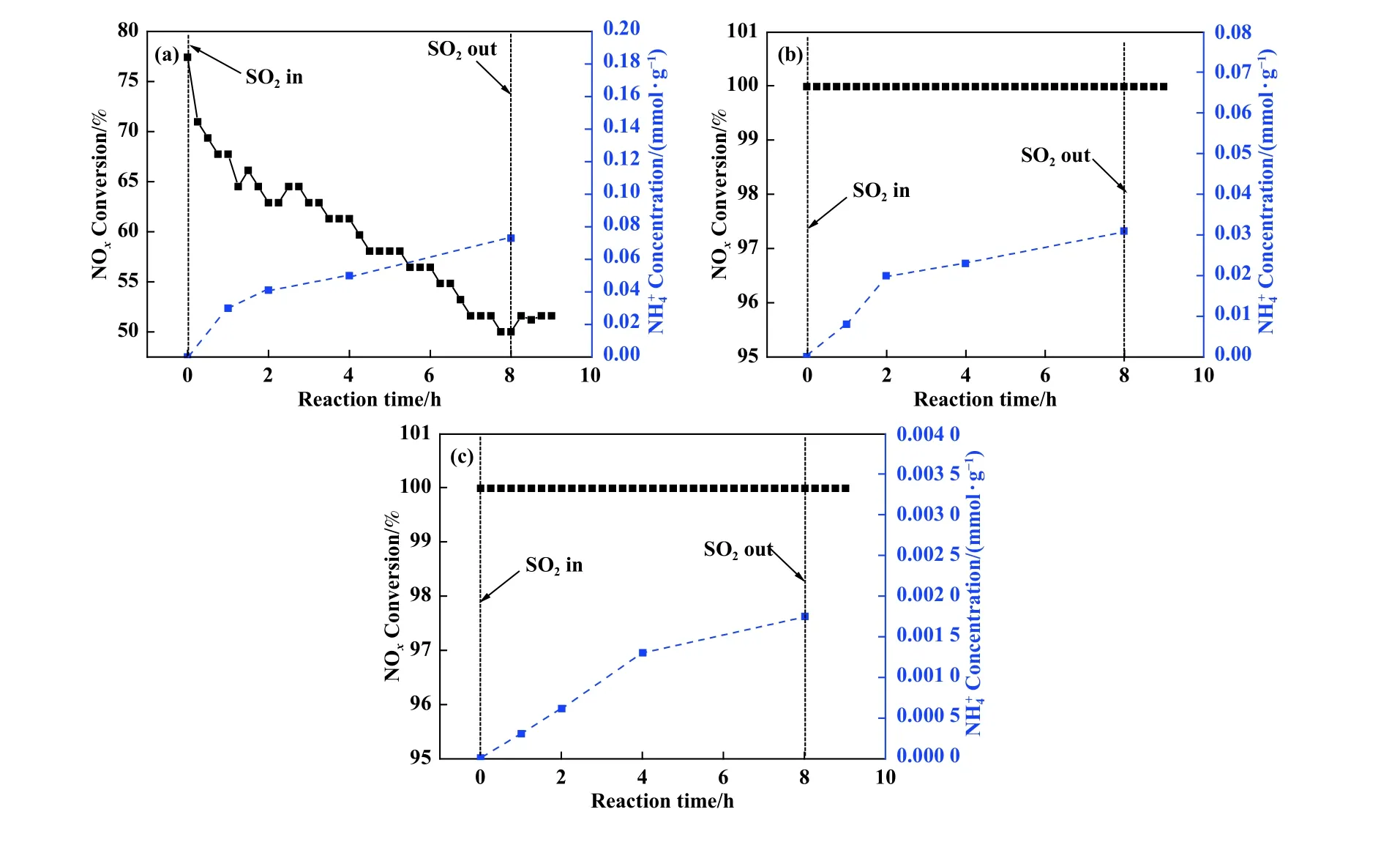
图4 不同温度中毒后V-Ce(0.1)/TiO2 催化剂水洗后的浓度(a) 180 ℃; (b) 240 ℃; (c) 300 ℃Fig.4 Concentration of SO2-poisoned V-Ce(0.1)/TiO2 catalysts at different temperatures(a) at 180 ℃; (b) at 240 ℃; (c) at 300 ℃
2.2.4 表相硫组分XPS 分析
为了进一步对中毒后催化剂表面化学性质进行表征, Ce、 V 元素的XPS 谱图如图5 所示, Ce 3d谱图可拟合出8 个特征峰, 其中标记为u'''/v'''、u''/v''和u/v 的双峰可以归结为Ce4+, 标记为u'/v'的双峰归结为Ce3+[19], 经过SO2中毒后, 可以发现随着反应温度升高, u'和v'峰逐渐凸显, 该结果表明随着反应温度升高催化剂表面的Ce3+占比逐渐增加.根据表2 中原子含量与价态比例结果, 可以发现新鲜VCe(0.1)/TiO2催化剂的Ce3+/Ce 比例为30.9%, 随着反应温度从180 升高至300 ℃, Ce3+/Ce 比例升高至32.3%、33.9%、 36.8%.Poston 等[17]研究了4 价、3 价硫酸铈的分解, 结果表明4 价硫酸铈的分解温度在700 ℃左右, 3 价硫酸铈则在800 ℃, 结合TGDTG 中760~930 ℃高温区间内的失重, 这里可以推断, 随着反应温度的升高, 催化剂表面逐渐生成较多的Ce2(SO4)3.V 元素的XPS 谱图如图5(b)所示,从图中可以看出, V 2p谱图中出现了两个特征峰,分别位于517.0、 515.8 eV, 分别归属于V5+和V4+.XRD 结果表明新鲜催化剂中存在Ce(Ⅲ)V(Ⅴ)O4这一晶相, XPS 谱图中Ce4+和V4+价态的出现表明了部分金属氧化物以高度分散的形式负载在催化剂上,未在XRD 谱图中出现衍射峰.表2 计算了V5+/V4+的比例, 从表中可以看出, 随着反应温度的升高,V5+/V4+比例逐渐降低, 这可能是由于高度分散的VO2在高温下被硫酸化, 生成大量的VOSO4.表2给出了S 元素的定量结果, 随着温度升高, 表面S含量逐渐增加, 这可能是由于高温下大量的金属氧化物被硫酸化, 导致表面S 元素含量大大增加.
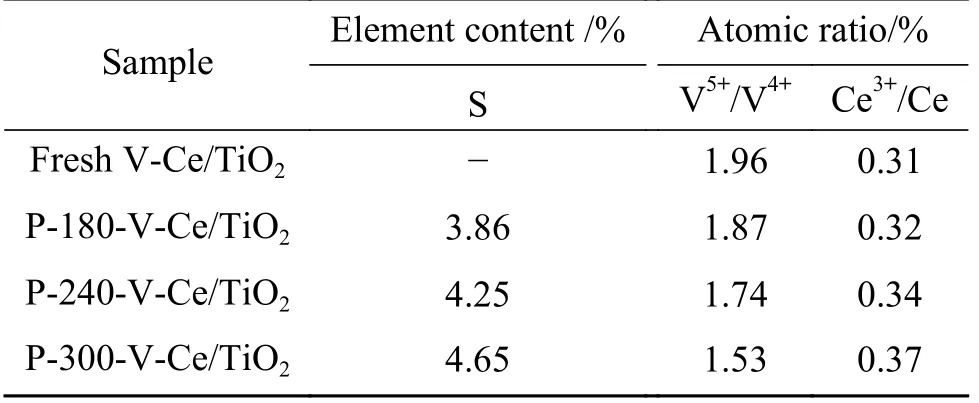
表2 不同温度中毒后V-Ce(0.1)/TiO2 催化剂表面原子含量及价态比例Table 2 Surface compositions of fresh and poisoned V-Ce(0.1)/TiO2 catalysts
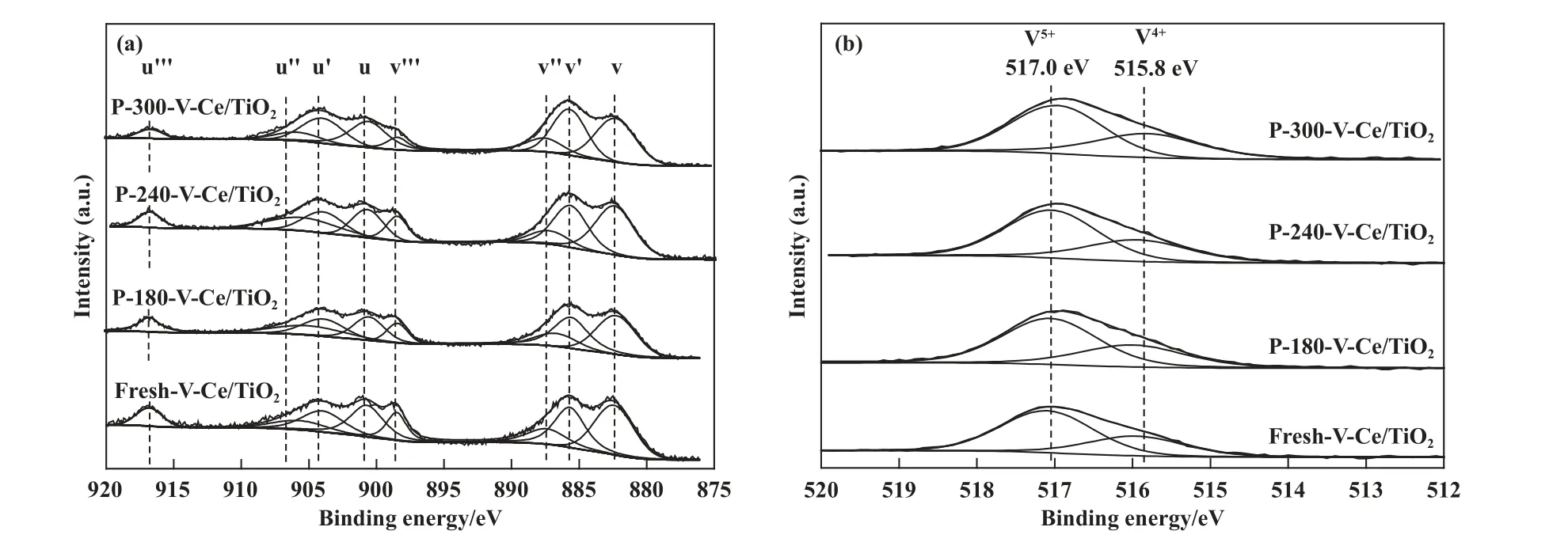
图5 不同温度中毒后V-Ce(0.1)/TiO2 催化剂的XPS 谱图(a) Ce 3d; (b) V 2pFig.5 XPS Pattern of V-Ce(0.1)/TiO2 catalysts after SCR reaction at different temperatures in SO2-containing atmosphere(a) Ce 3d; (b) V 2p
2.2.5 热再生后催化性能分析
为了进一步探究硫酸氢铵、 金属硫酸盐两种副产物对V-Ce(0.1)/TiO2催化剂失活的影响, 我们将催化剂上沉积的硫酸氢铵副产物剥离开来, 即对180 ℃中毒后催化剂进行550 ℃下N2气氛下的热再生, 并重新进行150~330 ℃无硫氛围下的脱硝活性测试, 以此确定两种硫组分对催化剂低温活性的损害主次地位.新鲜催化剂、 热再生前后催化剂的脱硝活性结果如图6 所示, 从图中可以看出两种硫组分的共同沉积导致150 ℃下活性从50.0%降至29.1%, 180 ℃下活性从77.8%降至51.2%, 210 ℃下活性影响较小, 从100%降至89.2%.随着热再生去除硫酸氢铵, 催化剂脱硝活性在150、 180 ℃下具有少量回升, 分别升至35.2%、59.5%.以上活性测试结果表明硫酸铵盐和金属硫酸盐的生成共同导致了180 ℃含硫氛围下V-Ce(0.1)/TiO2催化剂脱硝活性持续下降, 中毒后脱硝活性下降了26.6%, 其中硫酸氢铵的沉积导致了催化剂活性降低8.3%, 金属硫酸盐的沉积导致了催化剂活性降低18.3%.
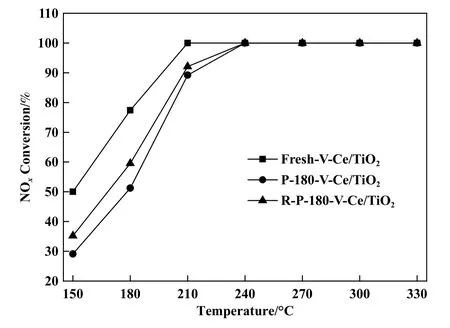
图6 热再生前后V-Ce(0.1)/TiO2 催化剂的脱硝活性结果Fig.6 The SCR performance of V-Ce(0.1)/TiO2 catalyst before and after thermal regeneration
2.2.6 NH3-TPD 和H2-TPR
为了进一步探究中毒前后催化剂的酸性、 氧化还原性能的变化, 我们对催化剂进行NH3-TPD 和H2-TPR 分析, 结果如图7 所示, 从图7(a)中可以看出, 新鲜催化剂主要表现处弱酸和强酸吸附位点, 脱附峰中心分别位于180 和600 ℃.经过180 ℃抗硫实验后, 催化剂的NH3脱附峰有所增强, 表明催化剂酸性有所增强, 随着温度升高至300 ℃, NH3脱附峰温度无明显变化, 脱附峰强度明显增强, 这可能是由于大量金属硫酸盐的生成增强了催化剂酸性, 导致NH3的吸附量明显增加.H2-TPR 谱图如图7(b)所示, 从图中可以看出, V-Ce(0.1)/TiO2催化剂的还原峰中心温度为465 ℃, 该还原峰归属于高度分散态的V2O5的还原, 随着反应温度的升高, 还原峰面积逐渐降低, 表明高温中毒后催化剂上可还原的V2O5大幅减少, 催化剂氧化还原性能大幅降低.
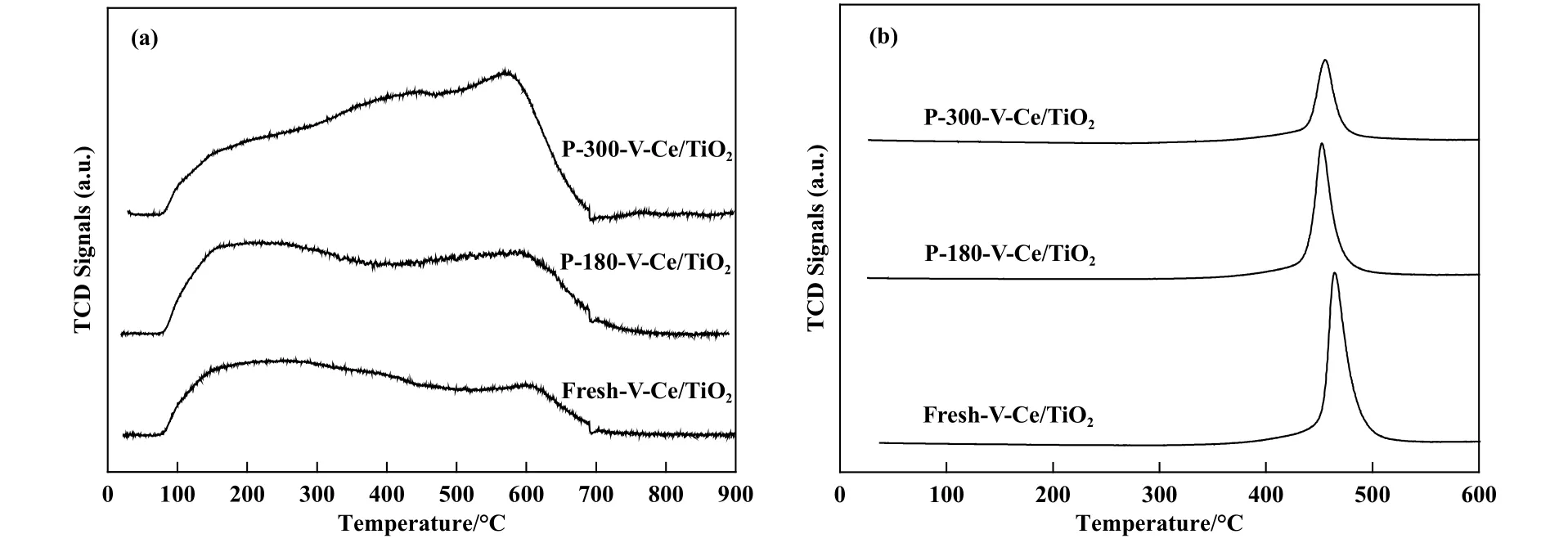
图7 V-Ce(0.1)/TiO2 催化剂中毒前后的NH3-TPD (a)和H2-TPR 图谱 (b)Fig.7 NH3-TPD Pattern (a) and H2-TPR pattern (b) of fresh and poisoned V-Ce(0.1)/TiO2 catalysts
2.3 原位DRIFTS
为了进一步探究金属硫酸盐的沉积对低温脱硝活性的影响, 我们对热再生后的V-Ce(0.1)/TiO2进行原位红外测试, 以探究催化剂反应路径的变化情况.图8(a)和8(b)是新鲜催化剂和热再生后催化剂预吸附NH3后与NO+O2反应的原位红外图谱, 从图中可以看出1 663、1 600、1 420 cm-1处出现了3个振动峰, 其中1 663 和1 420 cm-1处的振动峰可归结于吸附在B 酸位点上的, 1 600 cm-1处的振动峰可归因于NH3被晶格氧过度催化氧化而形成的硝酸盐中间产物[22].随着NO+O2的通入,1 420 cm-1处的振动峰强度明显减弱, 该结果表明吸附在新鲜V-Ce(0.1)/TiO2催化剂表面的NH3具有较高的反应活性.预吸附NO+O2饱和后进一步与NH3反应的原位红外图谱如图8(b)所示,从图中可以看出, 预吸附NO+O2饱和后催化剂表面出现了位于1 412 cm-1的振动峰, 该峰可归结于游离, 随着6 min 的NH3的通入, 该吸附峰峰强度未发生明显变化, 随着NH3的进一步通入,位于1 600 和1 420 cm-1处的NH3吸附峰开始出现, 表明新鲜催化剂上吸附的NO 物种不具有NH3-SCR 反应活性.以上结果表明新鲜催化剂的NH3-SCR 反应遵循ER 反应路径.中毒后并经过热再生处理后的催化剂的原位红外图谱如图8(c)和8(d)所示, 从图中可以看出相比于新鲜催化剂, 额外出现了1 288 cm-1处的倒峰和1 220 cm-1处的正峰, 分别可归属于吸附在在的S=O 键上的NH3和L 酸位上的NH3, 随着NO+O2的通入, NH3吸附峰峰强度仅少量减弱, 该结果表明热再生处理后催化剂表面吸附的NH3反应活性较低, NH3-SCR 反应的ER 反应路径被切断.预吸附NO+O2饱和后进一步与NH3反应的原位红外图谱如图8(d)所示, 吸附饱和后出现了位于1 420、1 254 cm-1处的负峰, 可归因于吸附在的S=O 键上的NO[23].随着8 min 的NH3通入, 位于1 254 cm-1的负峰完全消失, 1 420 cm-1处的吸附峰强度大幅降低.以上结果表明金属硫酸盐的存在增强了NO 的吸附和活化,NH3-SCR 反应遵循LH 反应路径.
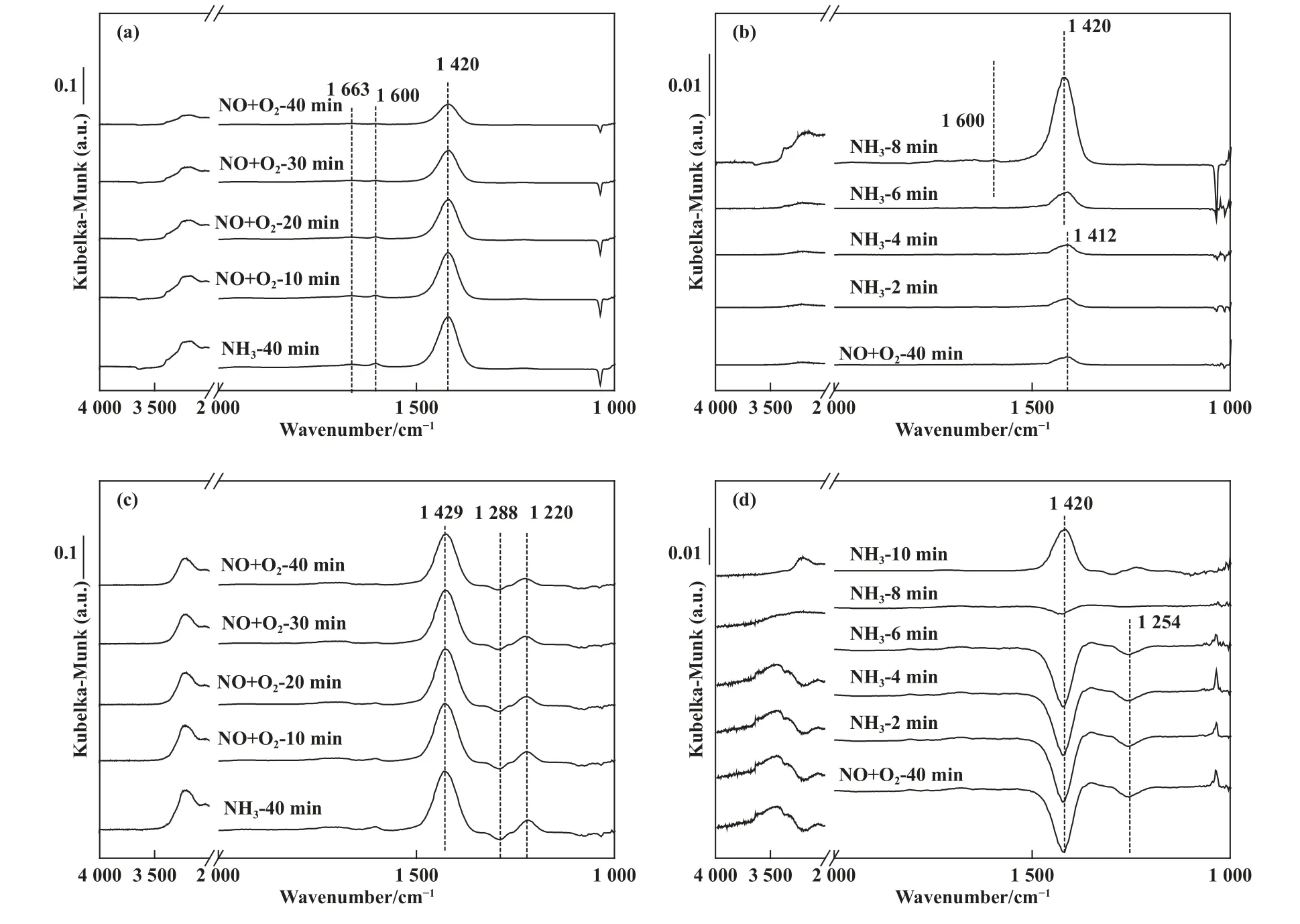
图8 中毒前后V-Ce(0.1)/TiO2 催化剂在180 ℃下预吸附气体后反应的原位红外图谱(a), (c) 为新鲜及中毒后催化剂先通入NH3 后通入NO+O2; (b), (d) 为新鲜及中毒后催化剂先通入NO+O2 后通入NH3Fig.8 DRIFTs Spectra of fresh and poisoned V-Ce(0.1)/TiO2 catalysts at 180 ℃(a), (c) reaction between NO+O2 and pre-adsorbed NH3 over fresh and poisoned catalysts;(b), (d) reaction between NH3 and pre-adsorbed NO+O2 over fresh and poisoned catalysts
2.4 V-Ce/TiO2 催化剂中低温抗硫活性小结
普遍认为, NH3-SCR 活性取决于催化剂的酸性、氧化还原性能, 其中表面酸性为NH3的吸附提供足够的吸附位点, 充足的氧化还原性能则有助于吸附态NH3的活化及进一步反应.如图1 所示, VCe/TiO2催化剂在180 ℃下经过8 h 抗硫中毒反应后, 活性从77.4%下降至50.0%, 随着反应温度升高至240、300 ℃, 催化剂活性保持100%不变.体相及表相硫组分分析结果表明, 180 ℃下金属硫酸盐、表面硫含量分别为3.21%(质量分数)、3.86%(原子分数), 300 ℃反应后金属硫酸盐、表面硫含量上升至4.45%(质量分数)、4.65% 原子分数).原位红外测试结果表明新鲜V-Ce/TiO2催化剂低温下遵循E-R反应机理, 吸附后的NH3能够被进一步活化并与气态NO 反应, 金属硫酸盐生成后催化剂低温下遵循L-H 反应机理, 且表面酸性明显增加.
为了进一步探究金属硫酸盐的NH3-SCR 活性,Ce(SO4)2/TiO2催化剂被制备并用于活性测试, 结果如图9 所示, 从图中可以看出, 180、300 ℃下NH3-SCR 活性分别为3.4%、24.6%, 该结果表明催化剂表面的金属硫酸盐在高温下具有NH3-SCR 催化活性.基于此, V-Ce/TiO2催化剂中低温抗硫性能差异可作如下总结: 低温NH3-SCR 反应需要较高的催化剂氧化还原性能, V-Ce/TiO2催化剂表面金属硫酸盐的生成则大大降低了氧化还原性能, 导致吸附态NH3无法活化并参与SCR 反应, 同时酸性的增强导致催化剂表面被大量NH3覆盖, NO 无法被同时吸附, 因此低温下E-R、L-H 反应路径均被切断; 中高温NH3-SCR 反应则需要较强的酸性, 金属硫酸盐的大量生成大大增强了V-Ce/TiO2催化剂酸性, 表相、体相中部分V2O5、CeO2的存在提供了所需的氧化还原性能, 因此V-Ce/TiO2催化剂的中高温抗硫活性能稳定维持在100%.
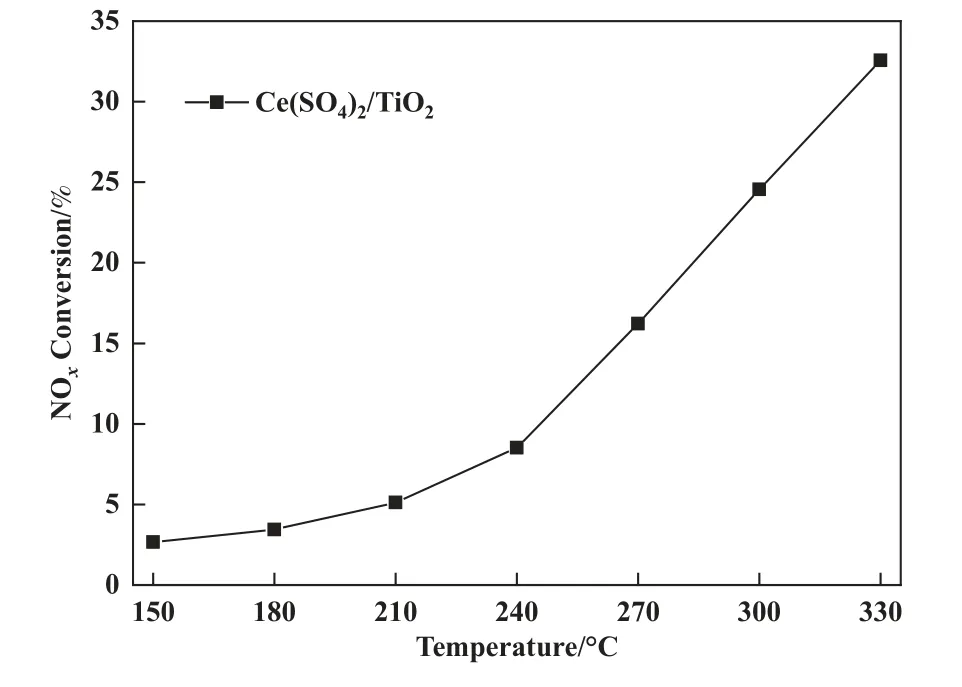
图9 Ce(SO4)2/TiO2 催化剂的脱硝活性Fig.9 NOx Conversion over Ce(SO4)2/TiO2 catalyst
3 结论
通过含硫氛围下的NH3-SCR 活性演变规律, 表征中毒后催化剂表相、体相硫组分, 结合原位红外测试表征NH3-SCR 反应机理, 本研究获得了VCe(0.1)/TiO2催化剂在180、240 和300 ℃下含硫氛围的NH3-SCR 反应中毒机理.结果表明, 180 ℃下催化剂上沉积了大量的硫酸氢铵和少量的金属硫酸盐, 共同导致在8 h 内活性从77.8%降至51.2%, 随着温度升高至300 ℃, 催化剂表面硫酸氢铵逐渐减少, 金属硫酸盐逐渐增多, 表面S 元素含量逐渐增多, 反应活性无明显下降.热再生后的活性测试结果表明180 ℃下硫酸氢铵的沉积导致了催化剂活性降低8.3%, 金属硫酸盐的沉积导致了催化剂活性降低18.3%.不同温度下的抗硫活性结果表明, 低温NH3-SCR 反应需要较高的氧化还原性能, 中高温NH3-SCR 反应则需要较高的酸性, 金属硫酸盐的生成导致了氧化还原性能降低、酸性增加, 因此低温NH3-SCR 活性大幅降低, 中高温活性则能保持在100%.
猜你喜欢
云南化工(2021年5期)2021-12-21 07:41:16
矿产综合利用(2020年1期)2020-07-24 08:51:20
中国化肥信息(2020年2期)2020-01-20 07:53:15
四川冶金(2019年5期)2019-12-23 09:04:48
中国化肥信息(2019年4期)2019-01-17 18:47:06
经济技术协作信息(2018年30期)2018-11-22 06:21:16
中学生理科应试(2017年2期)2017-04-01 00:05:46
中国资源综合利用(2016年1期)2016-02-03 02:55:12
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26 11:26:16
现代农业科技(2006年23期)2007-01-05 03:23:54
