
POMT1基因变异在α-抗肌萎缩相关糖蛋白病中的研究进展
2023-12-30 07:55石润茜徐盈常媛媛张建芳
新医学 2023年12期
关键词:沃克
石润茜?徐盈?常媛媛?张建芳

【摘要】 蛋白O-甘露糖基转移酶1(POMT1)基因编码的蛋白参与蛋白质O-甘露糖基化修饰起始步骤,在细胞连接、神经元迁移等多种生理过程发挥重要作用,α-抗肌萎缩相关糖蛋白病(α-DGP)是一组由α-抗肌萎缩相关糖蛋白(α-DG)O-糖基化缺陷所致的肌营养不良相关疾病。POMT1基因作为α-DGP的致病基因之一,通常与症状严重、预后差的α-DGP表型密切相关。该文对POMT1基因变异相关α-DGP的诊治、基因型-表型关系及可能的发病机制进行综述,以期深入探索POMT1变异的致病机制,为POMT1基因变异相关α-DGP的分子生物学水平治疗提供新思路。
【关键词】 先天畸形;沃克-沃伯格综合征;蛋白O-甘露糖基转移酶1基因;α-抗肌萎缩相关糖蛋白病;
蛋白O-甘露糖基转移酶;基因型-表型关系
Research progress on POMT1 gene variation in α-dystroglycanopathy Shi Runqian,Xu Ying, Chang Yuanyuan, Zhang Jianfang. Department of Gynecology and Obstetrics, the First Affiliated Hospital of Air Force Military Medical University, Xian 710032, China
Corresponding author, Zhang Jianfang, E-mail: zhzhhao@163.com
【Abstract】 Protein O-mannosyl-transferase 1 (POMT1) gene-encoded protein participates in the initial step of protein O-mannosylation modification and plays an important role in various physiological processes,such as cell connection and neuronal migration,etc. α-dystroglycanopathy (α-DGP) is a group of muscular dystrophy-related diseases caused by defects in α-dystroglycan (α-DG) O-glycosylation. As one of the causative genes of α-DGP,POMT1 gene is usually closely correlated with clinical phenotype of α-DGP with severe symptoms and poor prognosis. In this article,clinical diagnosis and treatment,genotype-phenotype relationship,and possible pathogenesis of α-DGP associated with POMT1 gene variation were reviewed,aiming to further explore the pathogenic mechanism of POMT1 variation and provide new ideas for the molecular biology-level treatment of POMT1 gene variation-related α-DGP.
【Key words】 Congenital abnormality; Walker-Warburg syndrome; POMT1 gene; α-dystroglycanopathy;
Protein O-mannosyl-transferase; Genotype-phenotype relationship
α-抗肌萎缩相关糖蛋白病(α-DGP)是一组罕见的肌营养不良相关疾病,由α-抗肌萎缩相关糖蛋白(α-DG)O-连接甘露糖基化缺陷导致。α-DGP是常染色体隐性遗传病,患者通常表现为肌肉、中枢神经系统及眼部受累。目前已明确19种致病基因,主要分为3类:第一类为编码抗肌萎缩相关糖蛋白(DG)核心区域,如DAG1基因;第二类为参与DG蛋白翻译后糖基化修饰的糖基化转移酶基因,如POMT1基因;第三类为糖基转移酶提供糖基化修饰底物的基因[1-2]。α-DG糖基化修饰是其与层粘连蛋白、轴突蛋白、基底膜聚糖等细胞外基质连接的必要反应,确保了细胞内外正常的物质交换和细胞信号转导[3]。POMT1基因参与α-DG糖基化修饰过程起始步骤。因此,POMT1致病性变异引起糖基化修饰缺陷严重,进而导致严重α-DGP临床表型。现对POMT1基因变异致病机制、与其相关α-DGP的临床特点及其基因型-表型关系等进行总结。
一、POMT1基因概述
POMT1基因位于9号染色体9q34.13,编码747个氨基酸,共有20个外显子,全长约21 kb。POMT1编码蛋白质O-甘露糖基转移酶1蛋白,该蛋白主要包括两类高度保守的15个结构域,即12個蛋白甘露糖基转移酶(PMT)跨膜结构域以及3个甘露糖基转移酶受体、三磷酸肌醇受体、兰尼碱受体(MIR)结构域见图1。POMT1蛋白是参与翻译后修饰的重要蛋白之一,定位于内质网,通过与蛋白O-甘露糖基转移酶2(POMT2)形成甘露糖基转移酶(POMT)复合物,在蛋白质O-甘露糖基化的初始过程共同催化O-甘露糖残基转至丝氨酸/苏氨酸末端[4]。POMT1蛋白在人类组织细胞中广泛分布,在胎儿大脑、骨骼肌、心肌、睾丸等组织中含量尤其丰富。目前,研究最广泛的POMT1蛋白作用底物为α-DG。α-DG定位于细胞膜,在中枢及周围神经系统、神经肌肉接头等重要结构高表达。高度甘露糖基化的α-DG介导细胞外基质与细胞骨架间的相互作用,对于维持肌肉和脑的正常结构有重要功能。
二、POMT1基因的变异类型及致病机制
已知的POMT1基因致病性变异包括无义变异、移码变异、剪接位点变异、框内删失/删失插入变异及错义变异,从纯合变异到复合杂合变异不等。
POMT复合物参与的O-甘露糖基化在维持细胞完整性、细胞形态方面发挥重要作用。糖基化是生物体内重要的翻译后修饰过程,通过促进蛋白质的正确折叠,从而提高蛋白质的稳定性。在小鼠胚胎早期发育中,POMT参与胚胎细胞间黏附连接形成,POMT1缺失对胚胎有致死性。果蝇POMT功能缺陷导致胚胎肌肉发育异常、感觉神经元轴突连接异常[5]。
在人骨骼肌中,甘露糖基化缺陷使细胞与细胞外基质中的层粘连蛋白2(laminin 2)的连接异常,进而使细胞骨架与肌动蛋白无法正常连接。在神经系统中,α-DG通过高度糖基化与具有层粘连蛋白球形结构域的胞外配体结合,从而维持神经元正常迁移等。放射状胶质细胞作为神经元移行的支架,在O-甘露糖基化不足时,尾足无法与基底膜锚定,导致神经元迁移过度或移行障碍,使脑皮质异常分层,这是α-DGP患者颅脑结构异常的重要原因之一。此外,有研究认为α-DGP患者智力发育迟缓与α-DG糖基化不足也密切相关。当小鼠神经元α-DG糖基化不足时,其海马的长期增益效应(LTP)被显著抑制,与之相应的学习记忆能力也显著降低[4]。然而,α-DG糖基化不足并不能解释全部临床表型[6]。既往研究发现,多种哺乳动物脑中细胞外基质为高度O-甘露糖基化,在小鼠视网膜感光细胞中发现存在其他O-甘露糖基化修饰蛋白,但具体分子及作用机制仍需进一步研究[7]。
三、α-DGP临床表现
α-DGP作为一组常染色体隐性遗传病,通常累及多个系统。其主要临床表现包括不同程度的中枢神经系统症状、肌无力,可伴眼部受累、关节畸形及癫痫发作等。患者智力、语言、运动能力发育通常受影响。临床表型根据起病时间、疾病进展及症状严重程度可分为沃克-沃伯格综合征(WWS)、肌-眼-脑病(MEB)、福山型先天性肌营养不良(FCMD)、先天性肌营养不良1C型(MDC1C)、先天性肌营养不良1D型(MDC1D)、先天性肌营养不良伴智力障碍(CMD-MR)、不伴智力障碍的肢带型肌营养不良(LGMD)或伴智力障碍的LGMD(LGMD-MR)[6]。
四、POMT1基因相关α-DGP的异质性
1. POMT1基因变异相关α-DGP临床表现
POMT1基因致病性变异导致POMT活性降低或丧失,进而使α-DG的糖基化合成减少,从而导致α-DGP的发生。截至2023年1月,人类基因突变数据库(HGMD)共收录99种疾病相关变异。与POMT1基因致病性变异相关α-DGP的临床表型主要包括有WWS、MEB、CMD-MR、LGMD。
WWS是α-DGP中最严重的表型,患者多在1岁内夭折,仅少数患儿可存活至3岁左右[5]。其特征性脑部受累表现为鹅卵石样无脑回畸形伴小脑或脑干发育不全。WWS患者发病年龄早,通常在出生或出生后短时间内出现进行性肌无力、肌张力低下、智力发育及运动能力严重低下,部分患者伴有癲痫发作。头颅MRI可显示Ⅱ型无脑回畸形、胼胝体缺失或发育不全、小脑及脑干发育异常、脑白质髓鞘异常、严重脑积水、枕骨脑膜膨出等。疾病累及眼部时,可出现高度近视、青光眼、白内障、水牛眼、小眼畸形、视网膜发育异常或脱落及视神经发育不良或萎缩等[8-10]。其他罕见症状包括男性外生殖器发育不良、肾积水以及唇腭裂等[11-12]。
MEB的临床表现与WWS部分重叠,但大多数患者疾病进展速度与表型严重程度轻于WWS,部分患者病情快速进展,平均生存年龄为18岁[9]。患者通常表现为肌力和肌张力低下伴有关节挛缩、眼部异常、智力低下,偶见癫痫发作。MEB患者眼部受累是其典型症状,易出现青光眼、青少年白内障、眼球震颤、进行性视网膜萎缩以及变性近视。此外,视觉诱发电位延迟出现且异常升高是其特征性表现。MEB患者颅内结构异常可表现为脑积水、鹅卵石样无脑回畸形、巨脑回、多小脑回、小脑轻度发育不全伴小脑囊肿、脑干扁平和扭折、脑桥发育不良及透明隔/胼胝体发育不全等[6]。
CMD-MR临床表型轻,通常无眼部畸形。患者肌无力症状出现较早,多数存在智力发育迟缓,可伴轻度头颅结构畸形,但畸形程度与智力障碍程度具有差异性。随患者年龄增长,智力可在一定程度有所提升,但仍低于正常水平[13]。
LGMD主要累及肢体近端的肌肉、关节。患者临床表现异质性大,起病时间晚,症状较轻,病情进展缓慢,通常在成年后丧失运动能力。LGMD主要包括LGMD2K、LGMD2N、LGMD2O等多种亚型。POMT1基因变异与LGMD2K亚型临床表现密切相关,早期可出现肢体近端肌无力进行性加重,半数患者可伴踝关节挛缩,60%患者合并有智力发育迟缓,部分患者可见小脑畸形、左心室扩张等症状[14-16]。
2. POMT1基因变异相关α-DGP基因型-表型关系
目前,普遍认为α-DGP表型严重程度与POMT活性呈负相关,POMT活性越高,临床表型越轻。通常当变异位点处于PMT/MIR结构域时,POMT活性受损程度更大,临床表型较严重[17-18]。
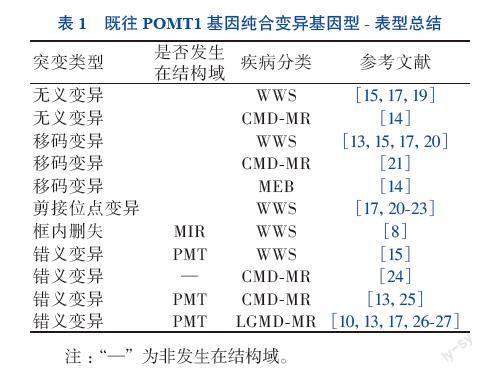
在POMT1基因纯合变异中,无论变异位点是否接近POMT1蛋白羧基端非结构域内,无义变异或移码变异均导致更严重的临床表型,如WWS、MEB等。错义变异表型相对较轻,但该变异发生在蛋白结构域内氨基酸高度保守区域时,也可出现WWS或MEB等严重表型。见表1。
POMT1基因复合杂合变异在POMT1基因相关α-DGP患者中更为多见,这类患者临床表型的严重程度与2个等位基因的变异类型及变异位点均密切相关。无义变异和(或)移码变异同时存在时,常表现为WWS。复合杂合变异中包括一种错义变异时,表型严重程度与错义变异位点、变异前后氨基酸理化性质异质性大小相关。错义变异位点处于结构域或高度保守区域时,临床表型相对更重,这可能由于发生在高度保守区域的错义变异会影响结构域正常构象及功能。见表2。
对于剪接位点变异,疾病严重程度与转录后mRNA保留的正常片段长度有关。剪接位点变异通常会导致重要外显子的缺失或移码变异,纯合变异表型通常更严重。若剪接位点变异及错义变异同时存在,需分析两者对表型及预后的共同影响。
五、POMT1变异相关α-DGP的诊断与治疗
目前,α-DGP的诊断方式主要包括测定患者的甘露糖基转移酶活性丧失程度、蛋白免疫印迹法检测α-DG低糖基化水平、出生后的临床症状及影像学表现以及全外显子测序(WES)技术等。随着WES技术在临床的广泛应用,其在明确α-DGP患者的基因诊断、提供遗传咨询、辅助产前诊断等方面发挥了重要作用[40-42]。对于有α-DGP患者的家庭,尽管患者父母在再次生育时可通过胎儿超声检查判断胎儿是否存在颅内结构异常,辅助诊断α-DGP,但该项检查至少在孕20周后方可进行。胚胎植入前单基因病遗传学检测能更早預防α-DGP再次发生。此外,体外培养患者真皮成纤维细胞,检测其POMT活性及糖基化程度,可能是准确评估低龄患者POMT1基因变异预后的方法之一[17]。
由于POMT1基因相关α-DGP患者表型及预后的异质性大,目前其临床治疗仍以个体化对症治疗、提高患者生活质量为主。大多数对症治疗仅限于维持肢体长时间的活动功能及预防并发症,如关节功能锻炼、跟腱挛缩松解、心肺功能早期筛查等。目前仍缺乏针对患者脑部畸形的有效疗法,尽管有手术治疗WWS患者的成功案例,但术后并发症及远期预后等问题不容忽视[8, 43]。此外,由于WWS患者在胚胎发育早期即存在神经元迁移异常,颅内解剖结构异常成为改善症状手术的难点[44]。随着基因组学的不断发展,更加精准的基因治疗方法被不断应用于临床。因此,需要进一步研究改善此类患者预后的相关疗法。
六、结 语
POMT1基因变异通常导致较严重的α-DGP表型。尽管部分POMT1基因错义变异的临床表型较轻,但多数患者智力发育依然受到严重影响。α-DG并非POMT复合物的唯一作用底物,因此探索其他作用底物及作用机制对解释临床表型异质性十分必要。由于α-DGP患者对家庭和社会有巨大的精神压力和经济负担,需要加强对高危人群的遗传咨询及产前诊断,降低α-DGP的发病率。随着分子生物学治疗手段逐渐运用于临床,探索POMT1相关α-DGP的基因治疗方法对改善患者症状及预后意义重大。
参 考 文 献
[1] Sheikh M O, Capicciotti C J, Liu L, et al. Cell surface glycan engineering reveals that matriglycan alone can recapitulate dystroglycan binding and function. Nat Commun, 2022, 13: 3617.
[2] Quereda C, Pastor ?, Martín-Nieto J. Involvement of abnormal dystroglycan expression and matriglycan levels in cancer pathogenesis. Cancer Cell Int, 2022, 22(1): 1-30.
[3] Sheikh M O, Halmo S M, Wells L. Recent advancements in understanding mammalian O-mannosylation. Glycobiology, 2017, 27(9): 806-819.
[4] 陈晓瑜, 熊晖. α-抗肌萎缩相关糖蛋白病脑结构畸形的研究进展. 生理科学进展, 2022, 53(1): 14-18.
[5] 甘思仪, 杨海燕, 肖婷, 等. POMT1和POMT2基因突变导致的2例α-抗肌萎缩相关糖蛋白病. 中南大学学报(医学版), 2021, 46(8): 915-919.
[6] 傅晓娜, 肖坤宏, 熊晖. α-抗肌萎缩相关糖蛋白病研究进展. 生理科学进展, 2019, 50(1): 10-18.
[7] Uribe M L, Martín-Nieto J, Quereda C, et al. Retinal proteomics of a mouse model of dystroglycanopathies reveals molecular alterations in photoreceptors. J Proteome Res, 2021, 20(6): 3268-3277.
[8] Kim D S, Hayashi Y K, Matsumoto H, et al. POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in α-DG. Neurology, 2004, 62(6): 1009-1011.
[9] Song D, Dai Y, Chen X, et al. Genetic variations and clinical spectrum of dystroglycanopathy in a large cohort of Chinese patients. Clin Genet, 2021, 99(3): 384-395.
[10] ?zyilmaz B, Kirbiyik ?, ?zdemir T R, et al. Impact of next-generation sequencing panels in the evaluation of limb-girdle muscular dystrophies. Ann Hum Genet, 2019, 83(5): 331-347.
[11] Bai L, Kovach A, You Q, et al. Structure of the eukaryotic protein O-mannosyltransferase Pmt1-Pmt2 complex. Nat Struct Mol Biol, 2019, 26(8): 704-711.
[12] Reynolds K, Zhang S, Sun B, et al. Genetics and signaling mechanisms of orofacial clefts. Birth Defects Res, 2020, 112(19): 1588-1634.
[13] Godfrey C, Clement E, Mein R, et al. Refining genotype-phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain, 2007, 130(10): 2725-2735.
[14] Al-Zaidy S A, Baskin B, Hawkins C, et al. Milder phenotype of congenital muscular dystrophy in a novel POMT1 mutation. Muscle Nerve, 2012, 45(5): 752-755.
[15] Bigotti M G, Brancaccio A. High degree of conservation of the enzymes synthesizing the laminin-binding glycoepitope of α-dystroglycan. Open Biol, 2021, 11(9): 210104.
[16] 羅苏珊, 卢家红. 肢带型肌营养不良. 中华神经科杂志, 2019, 52(7): 573-581.
[17] Geis T, R?dl T, Topalo?lu H, et al. Clinical long-time course, novel mutations and genotype-phenotype correlation in a cohort of 27 families with POMT1-related disorders. Orphanet J Rare Dis, 2019, 14(1): 179.
[18] Megarbane A, Bizzari S, Deepthi A, et al. A 20-year clinical and genetic neuromuscular cohort analysis in Lebanon: an international effort. J Neuromuscul Dis, 2022, 9(1): 193-210.
[19] Yis U, Uyanik G, Kurul S, et al. A case of Walker-Warburg syndrome resulting from a homozygous POMT1 mutation. Eur J Paediatr Neurol, 2007, 11(1): 46-49.
[20] Devisme L, Bouchet C, Gonzalès M, et al. Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain, 2012, 135(2): 469-482.
[21] Cotarelo R P, Fano O, Raducu M, et al. A double homozygous mutation in the POMT1 gene involving exon skipping gives rise to Walker-Warburg syndrome in two Spanish Gypsy families. Clin Genet, 2009, 76(1): 108-112.
[22] Finsterer J. Reply to:“Advances in imaging of brain abnormalities in neuromuscular disease”. Ther Adv Neurol Disord, 2019, 12: 175628641987832.
[23] Takeichi T, Nanda A, Aristodemou S, et al. Whole-exome sequencing diagnosis of two autosomal recessive disorders in one family. Br J Dermatol, 2015, 172(5): 1407-1411.
[24] Hafner P, Bonati U, Fischmann A, et al. Skeletal muscle MRI of the lower limbs in congenital muscular dystrophy patients with novel POMT1 and POMT2 mutations. Neuromuscul Disord, 2014, 24(4): 321-324.
[25] Messina S, Mora M, Pegoraro E, et al. POMT1 and POMT2 mutations in CMD patients: a multicentric Italian study. Neuromuscul Disord, 2008, 18(7): 565-571.
[26] Jacquemin V, Versbraegen N, Duerinckx S, et al. Congenital hydrocephalus: new Mendelian mutations and evidence for oligogenic inheritance. Hum Genomics, 2023, 17(1): 16.
[27] Alawneh I, Stosic A, Gonorazky H. Muscle MRI patterns for limb girdle muscle dystrophies: systematic review. J Neurol, 2023, 270(8): 3946-3957.
[28] Yang H, Manya H, Kobayashi K, et al. Analysis of phenotype, enzyme activity and genotype of Chinese patients with POMT1 mutation. J Hum Genet, 2016, 61(8): 753-759.
[29] Mohamadian M, Rastegar M, Pasamanesh N, et al. Clinical and molecular spectrum of muscular dystrophies (MDs) with intellectual disability (ID): a comprehensive overview. J Mol Neurosci, 2022, 72(1): 9-23.
[30] Marchuk M, Dovbonos T, Makukh H, et al. Sarcotubular myopathy due to novel TRIM32 mutation in association with multiple sclerosis. Brain Sci, 2021, 11(8): 1020.
[31] Yang H, Song D, Liu Y, et al. Seizures and EEG characteristics in a cohort of pediatric patients with dystroglycanopathies. Seizure, 2022, 101: 39-47.
[32] Chong Y K, Kwan Ma L C, Lo K L, et al. Dystroglycanopathy with two novel POMT1 mutations in a Chinese boy with developmental delay and muscular dystrophy. Eur J Paediatr Neurol, 2014, 18(4): 532-535.
[33] 陳晨, 梅世月, 朱朝锋, 等. 一个先天性肌营养不良症家系的POMT1基因突变鉴定与产前诊断. 中华医学遗传学杂志, 2018, 35(1): 78-80.
[34] Hu P, Wu S, Yuan L, et al. Compound heterozygous POMT1 mutations in a Chinese family with autosomal recessive muscular dystrophy-dystroglycanopathy C1. J Cell Mol Med, 2017, 21(7): 1388-1393.
[35] Johnson K, Bertoli M, Phillips L, et al. Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness. Skeletal Muscle, 2018, 8(1): 23.
[36] 乔凤昌, 胡平, 林颖, 等. 全外显子测序产前诊断Walker-Warburg综合征. 临床检验杂志, 2018, 36(5): 321-323.
[37] Bello L, Melacini P, Pezzani R, et al. Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy. Eur J Hum Genet, 2012, 20(12): 1234-1239.
[38] Carlson C R, McGaughey S D, Eskuri J M, et al. Illness-associated muscle weakness in dystroglycanopathies. Neurology, 2017, 89(23): 2374-2380.
[39] von der Hagen M, Becker L L, Wienker T F, et al. Just expect it: compound heterozygous variants of POMT1 in a consanguineous family: the role of next generation sequencing in neuromuscular disorders. Neuropediatrics, 2020, 51(1): 72-75.
[40] 吴若豪, 邱坤银, 唐文婷, 等. COL6A1基因嵌合突变的Bethlem肌病一例并文献复习. 新医学, 2021, 52(12): 941-946.
[41] 赵燕燕, 刘勇, 李乾. 原发性开角型青光眼家系的致病基因筛查. 眼科学报, 2020, 35(3): 161-166.
[42] 刘颖文,张玉鑫, 闫露露, 等. 指甲-髌骨综合征一家系的遗传学分析并文献复习. 新医学, 2023, 54(5): 365-369.
[43] Tanaka T, Harris C J, Barnett S S, et al. A successful treatment of endoscopic third ventriculostomy with choroid plexus cauterization for hydrocephalus in walker-warburg syndrome. Case Rep Neurol Med, 2016, 2016: 7627289.
[44] Ruan J, McKee K K, Yurchenco P D, et al. Exogenous laminin exhibits a unique vascular pattern in the brain via binding to dystroglycan and integrins. Fluids Barriers CNS, 2022, 19(1): 97.
(收稿日期:2023-05-17)
(本文编辑:林燕薇)
猜你喜欢
小品文选刊·印象大同(2022年7期)2022-05-30
知音海外版(上半月)(2022年4期)2022-05-14
科学之谜(2020年11期)2020-01-13
扣篮(2017年7期)2017-09-11
女士(2017年6期)2017-08-02
益寿宝典(2017年31期)2017-03-07
知音海外版(下半月)(2016年8期)2016-09-08
微型小说选刊(2015年9期)2015-11-17
女士(2015年6期)2015-05-30
知音海外版(上半月)(2015年1期)2015-05-14
