
矿物油中致癌多环芳烃测定方法对比及应用研究
2023-12-29 13:16:56秦红艳刘子楠栾利新赛尔达尼色依提郭鉴
润滑油 2023年6期
秦红艳,刘子楠,栾利新,赛尔达尼·色依提,郭鉴
(中石油克拉玛依石化有限责任公司,新疆 克拉玛依 834000)
0 引言
多环芳烃(PAHs)是指分子结构中含有两个或两个以上稠合芳香环的芳香烃,具有致畸、致癌、致突变、生物难降解的特性,是一种持久性有机污染物[1-3]。多环芳烃也是最早被发现的环境致癌物,部分多环芳烃经皮肤吸收和肺部吸入,在人体内不断累积,易诱发皮肤癌、肺癌和胃癌等严重疾病[4-7]。PAHs主要是由含碳化合物不完全燃烧或在石化燃料的使用过程中产生的,如煤炭、油、木头、垃圾和其他有机物质在不完全燃烧的情况下都会产生多环芳烃。而PAHs最初的来源是石油精炼过程中残留的焦油,因此煤焦油、石油、木炭、原油、柏油、矿物油、润滑油、电解溶液和染料中也可能会发现这些物质,橡胶制品中必不可少的原材料炭黑和填充油均含有多环芳烃[8-9]。

中石油克拉玛依石化有限责任公司是全世界单厂生产能力最大的高档环烷基润滑油生产企业,环烷基润滑油质量较好,是国内合成橡胶生产企业主要采用的填充油。中石油克拉玛依石化有限责任公司牵头建立国家标准GB/T 33322-2016《橡胶增塑剂芳香基矿物油》,要求多环芳烃采用SN/T 1877.3《矿物油中多环芳烃的测定方法》进行测定;2020年3月,国内轮胎橡胶行业牵头制定了GB/T 38529-2020 《轮胎中限用物质的限量要求》,提出了对制品、轮胎采用欧盟标准EN 16143进行测定[14]。环保是生产发展的趋势,从原理、前处理方式、适用范围和优缺点等方面对两种方法进行对比研究,对于企业生产、销售高黏度橡胶油填充油有指导意义。
1 国内外多环芳烃含量测定的主要方法
目前气相色谱-火焰离子化检测器(GC-FID)、气相色谱-质谱联用仪(GC/MS)和固相萃取-高效液相色谱紫外荧光串联(HPLC-UV/FL)是检测PAHs较常见的方法。针对PAHs检测的国家标准涉及燃料(GB/T 41071)、涂料(GB/T 36488)、动植物油脂(GB/T 24893、GB/T 23213)、水质(GB/T 13198、GB/T 26411)、热塑性弹性体(GB/T 29616)、化妆品(GB/T 29670)、电子电气产品(GB/T 29784)、硫化橡胶(GB/T 29614)、皮革(GB/T 36946)、炭黑(GB/T 3780.28)、鞋类(GB/T 33427、GB/T 33391)等,国际上涉及PAHs检测的有大气、饮用水、废水、固体废弃物、土壤、油品等。
矿物油中PAHs的检测标准目前有出入境检验检疫行业标准SN/T 1877.3和欧盟标准EN 16143。SN/T 1877.3采用液-液萃取原理,使用气相色谱/质谱联用(GC/MS)内标法定量,可测定16种多环芳烃(美国国家环境保护局规定的16种)。EN 16143主要采用硅胶层析法(双液相清洗)原理,使用气相色谱/质谱联用法(GC/MS)进行鉴定和定量,对于8种单独的 PAHs(REACH法规中规定的8种),方法的适用范围约为4~15 mg/kg,苯并[a]芘(BaP)的适用范围约为0.5~2 mg/kg。
2 两种方法测定油品中多环芳烃的比较
2.1 仪器与试剂
SN/T 1877.3和EN 16143均可采用气相色谱-质谱仪、PAHs混合标准溶液。SN/T 1877.3采用十二氘代苝(Peryline-di2)为内标物,纯度≥99%;EN 16143采用苯并[a]蒽-d12、苯并[b]荧蒽-d12和苯并[a]芘-d12为内标物,纯度> 98%;十氟联苯或氘化/C13标记的PAHs为注射标准物。
2.2 样品前处理方式
SN/T 1877.3方法测定样品中的PAHs是采用液-液萃取原理,样品先用环已烷溶解,用二甲基亚砜萃取后,加入氯化钠溶液降低二甲基亚砜提取液的溶解度,再用环己烷反萃取,将多环短侧链的芳烃萃取到环己烷层中。EN 16143主要采用硅胶层析法(双液相清洗),分离原理是根据物质在硅胶上的吸附力不同而得到分离。极性较大的物质与硅胶作用强,保留时间长。极性弱的物质与硅胶作用弱,保留时间短,物质在固定相与流动相间通过反复的吸附、解吸过程,得以分离。两种方法分析流程如图1所示。
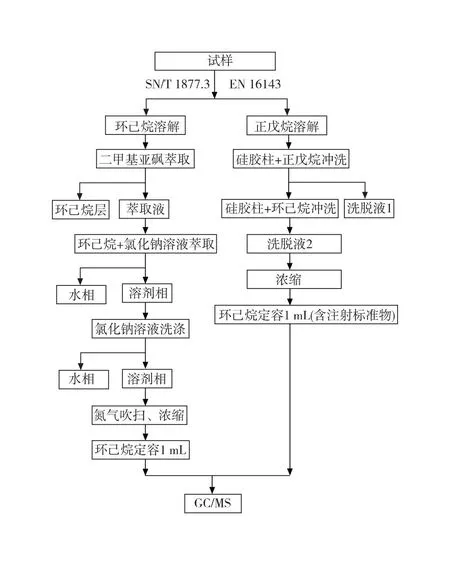
图1 SN/T 1877.3和EN 16143方法测定多环芳烃分析流程
SN/T 1877.3所用萃取试剂二甲基亚砜常温下为液体,熔点在18 ℃左右,低温下很容易结晶,因此试验温度至少20 ℃以上,才不至于因为环境对结果产生影响。此外,部分精制程度浅、胶质含量较高的样品,在萃取的过程中会发生乳化现象,需要进行破乳处理。
EN 16143方法中双液相清洗用正戊烷洗脱是为了将干扰测定结果的有机质除去,如:油品中的饱和组分、大部分极性较弱的芳烃等,而目标物采用环己烷洗脱,得到油品中多环短侧链的芳烃。在这个过程中,如果硅胶极性太强,正戊烷未能将干扰物洗脱,或者硅胶极性太弱,正戊烷将目标化合物也洗脱了,均会影响测定结果。EN 16143方法采用硅胶柱对样品清洗净化,方法只要求使用高纯度极硅胶,没有对使用的硅胶提具体要求,所以,在实际试验过程中要经过多次试验进行摸索,对使用的硅胶的规格和活化过程作统一。而且使用正戊烷、环己烷冲洗时分几部分将溶液加入硅胶柱,每次用量、倒入的速度、硅胶柱的长度都会影响目标物的洗脱速度。如若用量多倒入过快会因冲力过大将目标化合物洗脱;如若硅胶柱填充过紧,可能会导致干扰物不能全部洗脱、试验时间过长等问题。
2.3 检测多环芳烃的种类
采用GC/MS测定16种多环芳烃及内标物,16种多环芳烃标样及内标物的典型选择离子色谱如图2所示。

1.萘;2.苊烯;3.苊;4.芴;5.菲;6.蒽;7.荧蒽;8.芘;9.苯并[a]蒽;10.;11.苯并[a]蒽-d12(内标物);12.苯并[b]荧蒽;13.苯并[k]荧蒽;14.苯并(b)荧蒽-d12(内标物);15.苯并[a]芘;16.苯并[a]芘-12d(内标物);17.苝-d12(内标物);18.茚并[1,2,3-c,d]芘;19.二苯并[a,h]蒽;20.苯并[g,h,i]苝。
GC/MS能够满足测定16种多环芳烃标样及内标物的要求。对同一矿物油样品采用两种方法进行测定,结果如表1所示。
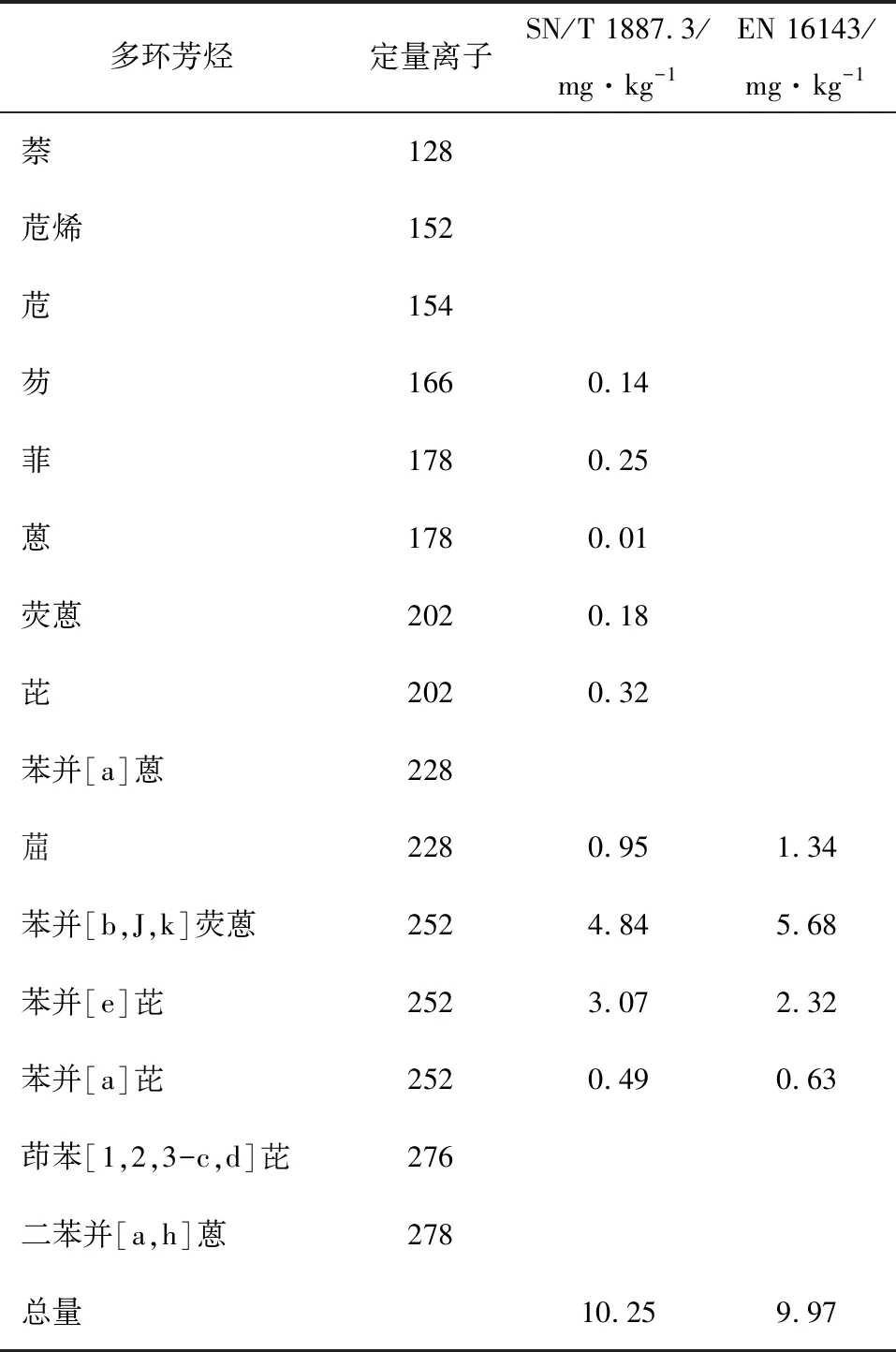
表1 两种方法测定结果对比
2.4 方法回收率试验对比
对SN/T 1887.3和EN 16143方法的加标回收率和相对标准偏差进行考察。重复测定6次,SN/T 1887.3和EN 16143加标回收率和相对标准偏差分别如表2和表3所示。

表2 SN/T 1887.3方法加标回收率及相对标准偏差(RSD)
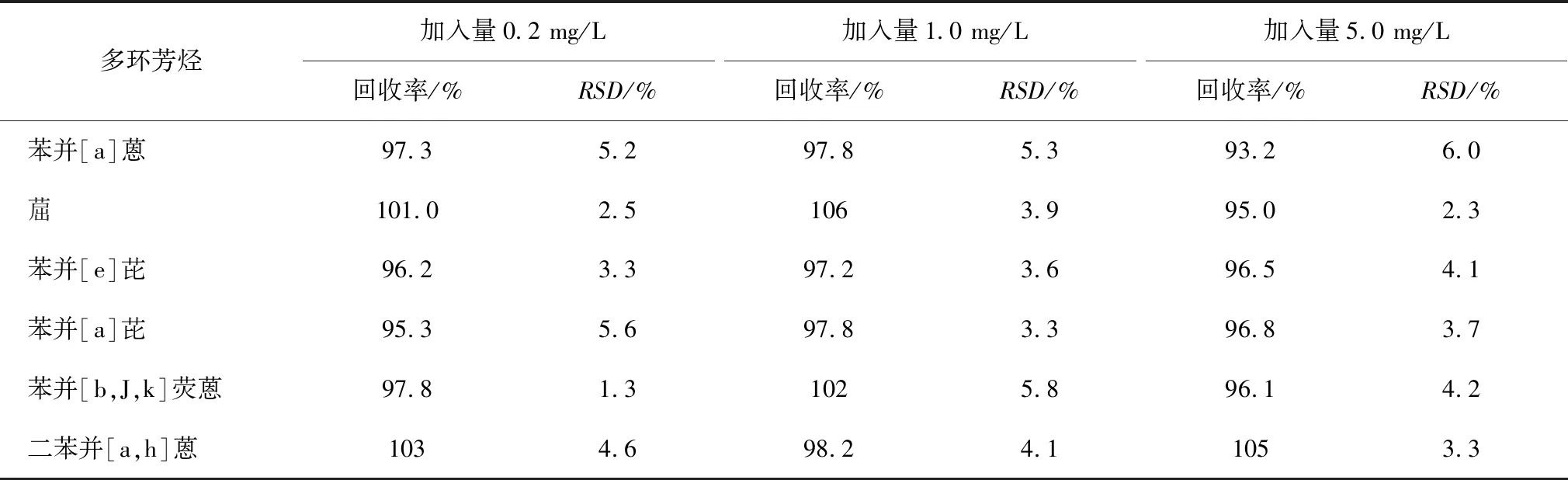
表3 EN 16143方法加标回收率及相对标准偏差(RSD)
SN/T 1887.3方法的整体加标回收率为75.3%~105.0%,相对标准偏差为2.2%~8.0%,由于前8种多环芳烃沸点相对较低,主要是因为净化过程(控制柱流速)及吹扫溶剂(吹扫时要控制流速、时间)操作复杂且时间长,容易引入测量误差,沸点较高的8种多环芳烃加标回收率为90.3%~105.0%。EN 16143方法测定的8种芳烃的加标回收率为93.2%~106.0%,略高于SN/T 1887.3方法测定结果,相对标准偏差为1.3%~6.0%。
3 结果与讨论
(1)SN/T 1887.3采用液-液萃取原理,考虑到萃取剂的性质,试验温度应控制在20 ℃以上;对于胶质含量较高的样品,在二甲基亚砜萃取前,先进行硅胶固相柱色谱净化除去胶质及其他杂环化合物;稠环芳烃含量较高的样品或环烷基矿物油,应适当降低称样量,将称样量控制在一个合理的范围,以排除干扰,提高结果的准确度。在对样品及结果进行全面评估后,可以考虑简化净化过程,以减少测量误差。
(2)EN 16143方法关键是调节固相吸附剂的极性,采用极性不同的洗脱剂(正戊烷、环己烷)将干扰物和目标物分离开。用加水失活的办法调节固相吸附硅胶的极性,需要通过试验确定最合适的加水量,避免影响硅胶活性。为了兼顾分离效果和分离效率,需确定适宜的硅胶柱的长度和粗细,并对加入的洗脱剂定次定量。
猜你喜欢
化工设计通讯(2022年12期)2023-01-24 05:38:54
化工学报(2021年10期)2021-10-31 23:36:50
科学技术创新(2021年19期)2021-07-16 10:07:18
发明与创新(2020年5期)2020-05-06 11:19:30
发明与创新·大科技(2020年2期)2020-04-17 09:52:22
科学导报(2019年73期)2019-12-20 08:46:53
天然气技术与经济(2018年1期)2018-03-06 07:42:57
造纸化学品(2018年5期)2018-01-31 03:26:27
化工设计通讯(2017年6期)2017-03-02 18:29:14
科技与创新(2016年10期)2016-05-28 03:17:18
