
花旗松素磷脂复合物白蛋白纳米粒的构建及其肠吸收研究
2023-12-25 13:19张佳慧孙敬蒙张诗雨张炜煜
中草药 2023年24期
张佳慧,孙敬蒙,张 鑫,张诗雨,张 欣,张炜煜*
1. 长春中医药大学药学院,吉林 长春 130117
2. 吉林大学第一医院 临床药学部,吉林 长春 130061
花旗松素又名二氢槲皮素、紫杉叶素,是一种二氢黄酮醇类化合物,为国家卫生健康委员会发布的2021 年第5 号文件批准的新食品原料;广泛存在于水飞蓟、落叶松、土茯苓和黄杞叶等50 多种植物体内[1]。花旗松素具备多种生物学活性,如抗氧化[2]、抗病毒[3]、抗炎[4]、抗肿瘤[5]等,具备很好的医药开发潜能,美国、俄罗斯、日本等国家以花旗松素为原料开发出多种药用、食用产品。但该成分水溶性低[6]、脂溶性差[7],肠道通透性差,导致口服吸收受限,从而影响临床疗效,限制其开发应用。现代研究开发了多种新型给药制剂,用于增强花旗松素的溶解度,包括脂质体[8]、纳米粒[9]、包合物[10]等。然而,仅提高其溶解度没有改善其渗透性,仍不能满足其功效发挥。因此,构建新型给药系统,协同改善其溶解性和膜渗透性,提高花旗松素体外溶出及体内吸收程度是当前口服给药途径研究的关键问题。
磷脂复合物白蛋白纳米粒是一种新型二元给药系统,能够提高药物的水溶性、脂溶性及体外溶出和体内吸收情况,而且磷脂和白蛋白均属于食品级辅料,因此将花旗松素制备成磷脂复合物白蛋白纳米粒,能提高花旗松素的功效。磷脂作为人体细胞膜的重要组成成分,能够提高药物与机体内胃肠道黏膜的生物相容性与亲和力,增加渗透性和滞留效应[11]。特别是天然磷脂,能够直接从大豆或蛋黄中提取,生物安全性良好[12]。白蛋白属于高度水溶性蛋白质,空间上具有网状结构和疏水区域,且携带多种药物结合位点,便于包载疏水性药物,具备提高药物溶解度的独特优势[13]。通常自牛血清、人血清或重组技术中获得,其中牛血清白蛋白(bovine serum albumin,BSA)凭借其丰富性、低成本、易纯化等特性被制药行业广泛接受。本研究首次将花旗松素载入磷脂复合物白蛋白纳米粒中,并采用Box-Behnken 设计(Box-Behnken design,BBD)-响应面法(response surface method,RSM)优化花旗松素磷脂复合物白蛋白纳米粒(taxifolin phospholipid complex albumin nanoparticles,Tax-PC/BSA NPs)的制备工艺,并对其进行质量评价;同时,通过大鼠在体单向肠灌流模型来评价Tax-PC/BSA NPs 的肠道吸收特性,为花旗松素制剂的进一步研发和应用提供参考。
1 仪器与材料
1.1 仪器
Agilent 1200 型高效液相色谱仪,安捷伦科技有限公司;TGL-18C 型高速台式离心机,上海安亭科学仪器厂;Scientz-II D 型超声波细胞粉碎机,宁波新芝生物科技股份有限公司;RE-52AA 型旋转蒸发器,上海亚荣生化仪器厂;Epsilon 2-4 LSC plus 型冷冻干燥机,德国Christ 公司;Infinite M200 Pro 型酶标仪,瑞士Tecan 公司;DSC3 型差示扫描量热(differential scanning calorimetry,DSC)仪,瑞士Mettler Toledo 公司;FTIR-8400S 型傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FT-IR)仪,日本岛津公司;XRD-7000 X 型射线衍射仪,日本Shimadzu 公司;H-7650 型透射电子显微镜(transmission electron microscope,TEM),Hitachi 公司;Zetasizer Nano ZS 型动态光散射仪,英国Malvern 公司;BS100-1A 型基本型蠕动泵,保定思诺流体科技有限公司;DKZ-2 型电热恒温振荡水槽,上海精宏实验设备有限公司。
1.2 药品与试剂
花旗松素对照品,批号111816-201102,HPLC测定质量分数≥98%,购自中国食品药品检定研究院;花旗松素原料药,批号20191208,质量分数≥90%,购自吉林健维天然生物科技有限公司;大豆卵磷脂(批号S30870,质量分数≥90%)、BSA(批号S12012,生物试剂,质量分数98%)、胃蛋白酶(批号S10027)、胰蛋白酶(批号S23119)、磷酸二氢钾(批号S24278,分析纯,质量分数99%)、磷酸氢二钾(批号S24279,分析纯,质量分数99%)均购自上海源叶生物科技有限公司;甲醇、乙腈均为色谱纯,无水乙醇、二氯甲烷、正辛醇均为分析纯,水为纯化水。
1.3 实验动物
本研究所用实验动物为SPF 级雄性SD 大鼠,体质量为180~200 g,购自辽宁长生生物技术股份有限公司,动物生产许可证号为SCXK(辽)2020-0001,动物实验经长春中医药大学实验动物伦理委员会批准,批准文号2023191。
2 方法与结果
2.1 花旗松素磷脂复合物(taxifolin phospholipid complex,Tax-PC)的制备
采用溶剂挥发法制备Tax-PC。取适量花旗松素和大豆卵磷脂溶于有机溶剂,恒温磁力搅拌一段时间后,旋转蒸发除去有机溶剂,加入适量二氯甲烷复溶,抽滤,减压干燥,即得Tax-PC。
2.2 Tax-PC/BSA NPs 及其冻干粉的制备
经过前期实验筛选,采用新型白蛋白纳米粒制备技术(NabTM技术)制备Tax-PC/BSA NPs。取Tax-PC 以二氯甲烷溶解,得油相;取处方量的BSA 加入纯水溶解,得水相;将油、水两相混合,采用超声波细胞粉碎仪超声一定时间,旋转蒸发去除有机溶剂,即得Tax-PC/BSA NPs。同样方法制备不含药物的空白PC/BSA NPs。将Tax-PC/BSA NPs 置于−80 ℃冰箱中预冻24 h,放入冷冻干燥机(温度<−40 ℃,真空度<20 Pa)36 h 后取出,称定质量,即得Tax-PC/BSA NPs 冻干粉。同样方法制备不含药物的空白PC/BSA NPs 冻干粉。
2.3 花旗松素含量测定
2.3.1 色谱条件 以Agilent Zorbox SB-C18柱为色谱柱;乙腈-水(22∶78)为流动相;检测波长290 nm;柱温30 ℃;进样量10 µL;体积流量1.0 mL/min。
2.3.2 对照品溶液的制备 取花旗松素对照品2.5 mg 置于25 mL 量瓶内,甲醇溶解并定容,摇匀,0.22 μm 微孔滤膜滤过,即得含花旗松素0.1 mg/mL的对照品溶液。
2.3.3 供试品溶液的制备 取Tax-PC/BSA NPs 混悬液5 mL,置于25 mL 量瓶内,加甲醇溶解并定容,摇匀,0.22 μm 微孔滤膜滤过,得供试品溶液。同法制备空白溶液。
2.3.4 专属性考察 取适量对照品溶液、供试品溶液及空白溶液,按“2.3.1”项下方法进行分析,记录色谱图,结果见图1。结果花旗松素色谱峰峰形良好,且测定不受辅料和溶剂的干扰,专属性良好。

图1 花旗松素对照品溶液 (A)、供试品溶液 (B) 和空白溶液 (C) 的HPLC 图Fig. 1 HPLC of taxifolin reference substance (A), test solution (B) and blank solution (C)
2.3.5 线性关系考察 精密吸取“2.3.2”项下对照品溶液适量,分别加甲醇稀释至质量浓度梯度为100、50、25、12.5、6.3、3.1 μg/mL,按“2.3.1”项下方法测定各质量浓度下样品含量,以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y),绘制标准曲线,得花旗松素的线性回归方程为Y=47.899X-0.035 1,R2=0.999 9,提示花旗松素在3.1~100.0 μg/mL 线性关系良好。
2.3.6 精密度试验 取“2.3.3”项下供试品溶液适量,分别加甲醇稀释至质量浓度梯度为75、50、25 μg/mL,在“2.3.1”项色谱条件下1 d 内测定5 次,日内精密度RSD 分别为1.75%、1.25%、1.42%,表明日内精密度良好;连续测定5 d,日间精密度RSD分别为1.42%、1.11%、0.85%,表明日间精密度良好。
2.3.7 稳定性试验 取“2.3.3”项下供试品溶液适量,分别在室温下放置0、2、4、6、12、24 h,按“2.3.1”项下色谱条件进样测定,结果花旗松素峰面积的RSD 为1.25%,表明稳定性良好。
2.3.8 重复性试验 按“2.3.3”项下方法制备供试品溶液6 份,按“2.3.1”项下色谱条件进样测定,结果花旗松素质量分数的RSD 为1.06%,表明重复性良好。
2.3.9 加样回收率试验 取“2.3.3”项下供试品溶液适量,分别加入低、中、高不同质量浓度的花旗松素对照品溶液,得到含药质量浓度梯度为80、100、120 μg/mL 的回收率样品溶液,按“2.3.1”项下色谱条件进样测定,计算加样回收率,结果花旗松素的回收率在97%~101%,平均加样回收率为99.22%,RSD 为0.82%,表明该方法符合检测要求。
2.4 复合率的测定
取Tax-PC 加甲醇超声溶解,稀释适当倍数,进样测定,记录峰面积,按公式计算复合率。
m1为测定的花旗松素含量,m0为投药量
2.5 药脂比的筛选
根据Tax-PC 的复合率,对不同药脂比2∶1、1∶1、1∶2、1∶3、1∶4 进行筛选。结果如表1 所示,随磷脂占比增高复合率逐渐增大,当药脂比达1∶3 后,复合率增长减缓。观察各比例下的TAXPC,磷脂用量增大,复合物干燥效率降低,且易出现黏结现象。综合考虑确定1∶3 为最佳药脂比。
表1 药脂比的筛选结果 (±s, n = 3)Table 1 Screening results of drug-phosphorus ratio (±s,n = 3)

表1 药脂比的筛选结果 (±s, n = 3)Table 1 Screening results of drug-phosphorus ratio (±s,n = 3)
与药脂比1∶3 组比较:**P<0.01**P < 0.01 vs drug to lipid ratio 1:3 group
药脂比 复合率/% 药脂比 复合率/%2∶1 60.72±0.28** 1∶3 89.12±0.69 1∶1 70.83±0.60** 1∶4 89.69±0.69 1∶2 84.60±0.18**
2.6 包封率、载药量和渗漏率的测定
经前期实验考察,采用高速离心法,设定条件离心转速14 000 r/min,离心时间20 min 进行Tax-PC/BSA NPs 的包封率、载药量和渗漏率测定,在该条件下纳米粒和游离药物得到了有效分离,同时不影响纳米粒结构稳定性。取Tax-PC/BSA NPs 1 mL于超滤离心管,甲醇破乳并定容,摇匀,0.22 µm 微孔滤膜滤过,测定包封与未包封的花旗松素总药量(m);取Tax-PC/BSA NPs 1 mL,置EP 管中,14 000 r/min 离心20 min,取上清液200 μL 于2 mL 量瓶,甲醇定容,测定游离药量(m游),进样测定含量,按公式计算包封率和载药量。
m纳为Tax-PC/BSA NPs 冻干粉质量
分别取新鲜制备的及置于4 ℃冰箱内储存48 h 后的Tax-PC/BSA NPs 混悬液,在14 000 r/min 条件下离心20 min,按公式计算纳米粒渗漏率。
m48为经48 h 沉降后的Tax-PC/BSA NPs 混悬液药物含量,m游为游离药量,m为投药量
2.7 BBD-RSM 优化Tax-PC/BSA NPs 的处方工艺
2.7.1 BBD 实验设计与结果 以总评归一值(OD)作为工艺优化的评价指标。采用数学转换计算归一值(d),其中渗漏率取值小为优,包封率和载药量取值大为优。公式分别如下。
d1、d2、d3分别为Tax-PC/BSA NPs 的包封率、载药量及渗漏率的归一值,Y1为实测值,Ymax、Ymin分别是包封率、载药量及渗漏率的实测最大值和最小值
将各指标的d值按下列公式计算,得OD 值。
在单因素实验的基础上,选择药/BSA 比(X1)、油水比(X2)、超声时间(X3)进行3 因素3 水平的分析实验。BBD 实验设计的因素与水平、安排与结果见表2。

表2 BBD 实验设计的安排与结果Table 2 Arrangements and results of BBD experimental design
2.7.2 BBD 实验设计数据分析 对表2 数据进行统计分析,通过拟合得二元多项回归方程为OD=0.768 0+0.206 2X1+0.058 8X2-0.062 5X3-0.002 5X1X2+0.040 0X1X3-0.225 0X2X3-0.132 8X12-0.242 8X22-0.300 3X32,R2=0.990 2。对回归方程进行方差分析,结果如表3 所示,所建立的回归模型具有显著性(P<0.01),表明该模型具有统计学意义。失拟项无显著性(P>0.05),说明模型拟合度良好,可用于分析和预测最优工艺。采用软件绘制等高线图和三维响应面图,结果见图2。最终通过软件分析优化得到最优处方为药/BSA 比1∶9.39,油水比1∶11.51,超声时间7.76 min。

表3 BBD 实验设计的方差分析结果Table 3 ANOVA results of BBD experimental design

图2 各因素间影响OD 值的响应曲面及等高线图Fig. 2 Response surface and contour map of various factors affecting OD value
2.8 Tax-PC/BSA NPs 最优处方工艺的验证
根据响应面实验结果,按照最优工艺平行制备3 批Tax-PC/BSA NPs 混悬液,结果如表4 所示,3批Tax-PC/BSA NPs 混悬液平均OD 值为0.82,与预测OD 值0.85 相对偏差较小,预测性良好,综上表明Tax-PC/BSA NPs 最优制备工艺稳定可靠。

表4 验证试验考察结果Table 4 Results of validation test investigations
2.9 Tax-PC/BSA NPs 的表征
2.9.1 TEM 观察 分别取空白PC/BSA NPs 与Tax-PC/BSA NPs 溶液,滴加于覆盖碳膜铜网上,以2%磷钨酸染色3 min,干燥后于TEM 下观察,可见空白PC/BSA NPs 和载药纳米粒(Tax-PC/ BSA NPs)均为形态规整的类球形,且无黏连聚集现象(图3)。

图3 空白纳米粒 (A) 和载药纳米粒 (B) 的TEM 图Fig. 3 TEM of blank nanoparticles (A) and drug-loaded nanoparticles (B)
2.9.2 粒径、PDI 与ζ 电位测定 取Tax-PC/BSA NPs 及空白纳米粒溶液适量,置于动态光散射仪样品池中,测定其粒径、ζ 电位以及PDI。结果显示,空白PC/BSA NPs 的平均粒径为(158.00±1.55)nm、PDI 为0.190±0.010 及ζ 电位为(−32.60±0.35)mV,Tax-PC/BSA NPs 的平均粒径为(184.90±0.98)nm、PDI 为0.275±0.010 及ζ 电位为(−36.60±0.53)mV,见图4。

图4 空白PC/BSA NPs (A) 和Tax-PC/BSA NPs (B) 的粒径分布 (I) 和ζ 电位 (II)Fig. 4 Particle size distribution (I) and ζ potential (II) of blank PC/BSA NPs (A) and Tax-PC/BSA NPs (B)
2.9.3 DSC 取花旗松素、大豆卵磷脂、BSA、物理混合物、TAX-PC、空白PC/BSA NPs 冻干粉、Tax-PC/BSA NPs 冻干粉各5.0 mg 置铝坩埚中,以空白坩埚为参比,载气为N2,体积流量50.0 mL/min,升温速度10 ℃/min,温程30~250 ℃,进行DSC分析。如图5 所示,大豆卵磷脂在129.17 ℃和180.17 ℃各存在2 个低强度、宽吸热峰,表明磷脂极性区域的熔化。TAX-PC 中花旗松素的熔点峰明显消失,而在174.50 ℃和253.83 ℃存在2 处低强度吸热峰,显示了药物的非晶化。说明花旗松素与大豆卵磷脂之间存在诸如氢键作用或范德华力等弱分子间相互作用,使两者结合形成磷脂复合物。BSA的熔点峰分别为94.31、220.50 ℃,花旗松素在231.25 ℃处有1 个强吸热峰,温度升高熔融了物理混合物,形成部分原位混合物,与原始峰相比,峰强度降低,表明物理混合物具有相加特性,该结果与Telange 等[14]报道一致。而Tax-PC/BSA NPs 冻干粉的DSC 曲线中,花旗松素的熔点峰被掩盖,且出现1 处新的大宽峰,峰值为78.83 ℃,表明了Tax-PC/BSA NPs 的形成。

图5 花旗松素、大豆卵磷脂、BSA、物理混合物、空白PC/BSA NPs、Tax-PC/BSA NPs 的DSC 谱图Fig. 5 DSC spectrum of taxifolin, soybean lecithin, BSA,physical mixture, blank PC/BSA NPs, and Tax-PC/BSA NPs
2.9.4 FT-IR 分析 采用KBr 压片法,将花旗松素、大豆卵磷脂、BSA、物理混合物、Tax-PC、空白PC/BSA NPs 冻干粉、Tax-PC/BSA NPs 冻干粉分别与KBr 以1∶100 比例研匀,压片进行测定。观察图6发现,物理混合物展现了物质的单纯加和性;花旗松素中,3 549.02、3 402.43 cm−1属于-OH 吸收特征峰;大豆卵磷脂中,2 924.08、2 852.71 cm−1为脂肪酸酯-CH2-的不对称振动吸收峰,1 230.58、1 058.91 cm−1为大豆卵磷脂的P=O 基团的伸缩振动峰。Tax-PC 的-OH 峰明显消失,并在3 404.36 cm−1处显示1大宽峰,可能是大豆卵磷脂的P=O 基团与花旗松素的-OH 结合,致使-OH 峰变弱,并与大豆卵磷脂的3 396.64 cm−1附近的宽峰相重叠。因此,推测花旗松素与磷脂分子间可能通过氢键作用形成了Tax-PC。载药后的Tax-PC/BSA NPs 中花旗松素的羟基酚吸收峰(3 549.02、3 402.43 cm−1)、苯环骨架峰(1 585.48 cm−1)被掩盖,药物吸收特征峰消失,表明花旗松素被包裹在纳米粒中。

图6 花旗松素、大豆卵磷脂、BSA、物理混合物、Tax-PC、空白PC/BSA NPs、Tax-PC/BSA NPs 的FT-IR 谱图Fig. 6 FTIR spectrum of taxifolin, soybean lecithin, BSA,physical mixture, Tax-PC, blank PC/BSA NPs, and Tax-PC/BSA NPs
2.9.5 XRD 分析 采用管流/管压(20 mA/40 kV),衍射范围3°<2θ<45°,扫描速度8°/min 的检测条件对花旗松素、大豆卵磷脂、BSA、Tax-PC、Tax-PC/BSA NPs 冻干粉进行XRD 分析。如图7 所示,花旗松素存在明显的晶体衍射峰,表明花旗松素是以结晶型存在的物质。而Tax-PC 中,花旗松素已由结晶型转为无定型,表明花旗松素与大豆卵磷脂结合形成了磷脂复合物。随着白蛋白纳米粒的进一步包封,物质依然显示无定型态,此形态有利于改善花旗松素的溶解度和释放速率。

图7 花旗松素、大豆卵磷脂、BSA、Tax-PC、Tax-PC/BSA NPs 的XRD 谱图Fig. 7 XRD spectrum of taxifolin, soybean lecithin, BSA,Tax-PC and Tax-PC/BSA NPs
2.10 理化性质测定
参考文献报道[15],采用摇瓶法测量溶解度和表观油水分配系数(lgP)。取过量花旗松素、Tax-PC、Tax-PC/BSA NPs 分别加入到纯化水、pH 1.2 盐酸溶液、pH 6.8 磷酸盐缓冲溶液(PBS)中,涡旋混合10 min,置37 ℃恒温振荡器中,100 r/min 连续振荡72 h,在3600 r/min 条件下离心10 min,取上清液加甲醇稀释适当倍数,滤过,进样测定,计算花旗松素平衡溶解度。另取适量花旗松素、Tax-PC、Tax-PC/BSA NPs 分别加入上述溶液的饱和正辛醇中,超声溶解、滤过(0.45 μm 微孔滤膜)得花旗松素正辛醇溶液。精密移取该溶液5 mL,与同体积正辛醇饱和纯化水及不同pH 值缓冲溶液混合,于37 ℃恒温水浴振荡24 h,静置,分别取上下层,进样测定,计算lgP。表5 结果显示,Tax-PC/BSA NPs在Tax-PC 的基础上,进一步提高了花旗松素的平衡溶解度,在水、pH 1.2 盐酸溶液、pH 6.8 PBS 中分别提高了38.48、32.72、48.31 倍。经白蛋白纳米粒进一步包封后的花旗松素,与磷脂复合物相比lgP值大于1 且基本持平,适宜于药物吸收,亲脂性仍得到保留。
表5 花旗松素、Tax-PC、Tax-PC/BSA NPs 的理化性质测定结果 (±s, n = 3)Table 5 Physical and chemical properties of taxifolin, Tax-PC and Tax-PC/BSA NPs (±s, n = 3)

表5 花旗松素、Tax-PC、Tax-PC/BSA NPs 的理化性质测定结果 (±s, n = 3)Table 5 Physical and chemical properties of taxifolin, Tax-PC and Tax-PC/BSA NPs (±s, n = 3)
样品 介质 溶解度/(mg∙mL−1) lgP花旗松素 水 0.888±0.009 0.79 pH 1.2 盐酸溶液 0.785±0.052 0.56 pH 6.8 PBS 1.433±0.005 0.75 TAX-PC 水 1.936±0.018 1.41 pH 1.2 盐酸溶液 1.743±0.002 1.12 pH 6.8 PBS 2.823±0.035 1.30 Tax-PC/BSA NPs 水 34.167±0.037 1.39 pH 1.2 盐酸溶液 25.684±0.003 1.15 pH 6.8 PBS 69.230±0.009 1.32
2.11 稳定性考察
2.11.1 储存稳定性 取Tax-PC/BSA NPs 冻干粉密封于西林瓶中,分别于4 ℃和室温放置3 个月,观察放置前后的外观,并测定放置前后渗漏率、粒径、PDI 和ζ 电位,以验证纳米粒稳定性。根据表6 可知,Tax-PC/BSA NPs 经4 ℃和室温条件下分别储存3 个月后各项指标变化程度均在可控范围,损伤较小,表明纳米粒冻干粉的储存稳定性良好,同时各指标结果均提示,纳米粒在4 ℃条件下储存优于室温,提示Tax-PC/BSA NPs 冻干粉应在4 ℃进行储存。
表6 纳米粒储存稳定性 (±s, n = 3)Table 6 Storage stability of nanoparticles (±s, n = 3)

表6 纳米粒储存稳定性 (±s, n = 3)Table 6 Storage stability of nanoparticles (±s, n = 3)
样品 粒径/nm ζ 电位/mV PDI 渗漏率/%储存前 184.90±0.98 −36.60±0.53 0.275±0.010 0.46±0.10 4 ℃储存3 个月 185.70±0.33 −36.00±0.24 0.280±0.020 0.49±0.08室温储存3 个月 188.10±0.08 −35.20±0.20 0.298±0.040 0.61±0.29
2.11.2 氧化指数 取Tax-PC/BSA NPs 溶于无水乙醇成澄明溶液,分别测定其在波长233 nm 及215 nm 的吸光度,计算氧化指数,3 批Tax-PC/BSA NPs的氧化指数值分别为0.179、0.179、0.181,均在0.2以下,符合脂质微粒制剂氧化程度。
2.12 体外模拟消化释放
参考文献报道[16],进行体外模拟消化释放。模拟消化液组成和消化条件如表7 所示,预先取3 mL花旗松素、Tax-PC 和Tax-PC/BSA NPs 样品溶液(均含花旗松素5 mg)分别与3 mL 模拟胃液(SGF)充分混合,调pH 至1.2,装入透析袋(MWCO3500),并置于存有150 mL 释放介质(乙醇与不含胃蛋白酶的SGF 按1∶1 等体积混合)的锥形瓶内,恒温振荡消化2 h 后完成。另取6 mL 模拟肠液(SIF)继续添加至含有上述混合物的透析袋中,调节pH 值至7.0,置于存有150 mL 释放介质(乙醇与不含胰蛋白酶的SIF 按1∶1 等体积混合)的锥形瓶内,恒温振荡消化4 h;全过程分别在10、30 min 及1、2、3、4、5、6 h 取适量释放介质,并补充同温度同量的新鲜介质,取样在4 ℃下保存。图8 结果显示,经2 h 胃部消化,花旗松素、Tax-PC/BSANPs 累积释放率分别为(20.56±0.35)%、(18.89±0.37)%、(11.21±0.65)%,胃部消化率均在25%以下,药物在胃部的吸收强度较低。转移至肠道消化环境模拟消化时(共4 h),花旗松素、Tax-PC、Tax-PC/BSANPs的肠道消化释放明显增强,花旗松素、Tax-PC、Tax-PC/BSANPs 最终累积释放率可达(48.26±0.71)%、(71.86±1.83)%、(82.73±0.62)%。

图8 花旗松素、Tax-PC、Tax-PC/BSANPs 在模拟胃-小肠消化液中的释放情况 (±s, n = 3)Fig. 8 Release of taxifolin, Tax-PC, Tax-PC/BSANPs in simulated stomach-small intestine digestive fluid (±s, n = 3)
2.13 大鼠在体肠吸收实验
2.13.1 供试品溶液的制备 Krebs-Ringer’s(K-R)试液:取氯化钙0.37 g、氯化钠7.8 g、氯化钾0.35 g、氯化镁0.02 g、葡萄糖1.4 g、碳酸氢钠1.37 g、磷酸二氢钠0.32 g,纯化水定容至1 L,摇匀,即得。
2.13.2 对照品溶液的制备 精密称取花旗松素对照品5 mg,置于50 mL 量瓶中,加适量K-R 试液溶解并定容,摇匀,0.22 μm 微孔滤膜滤过,即得花旗松素对照品溶液。
2.13.3 含药肠灌流供试液的制备 称取适量花旗松素、Tax-PC 及Tax-PC/BSA NPs 冻干粉,加入KR 试液配制成含花旗松素相同质量浓度的花旗松素、Tax-PC、Tax-PC/BSA NPs 肠灌流供试液。
2.13.4 空白肠灌流液 取37 ℃预热K-R 试液,进行在体肠灌流实验,循环2 h,收集灌流液,4 ℃冷藏备用。
2.13.5 专属性考察 取适量对照品溶液、含药肠灌流供试液及空白肠灌流液、按“2.3.1”项下色谱条件进行分析,记录色谱图,结果见图9。结果花旗松素色谱峰峰形良好,灌流液中的内源性物质对花旗松素无干扰,专属性良好。

图9 花旗松素对照品溶液 (A)、含药肠灌流供试液 (B) 和空白肠灌流液 (C) 的HPLC 图Fig. 9 HPLC of taxifolin reference substance (A), drugcontaining intestinal perfusion test solution (B) and blank intestinal perfusion solution (C)
2.13.6 线性关系考察 精密吸取一定量“2.13.2”项下对照品溶液,加入K-R 试液稀释成115、52、25、14、7、1.4 μg/mL,质量浓度的系列对照品溶液,按“2.3.1”项下色谱条件测定各质量浓度下样品含量,以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y),绘制标准曲线,得回归方程Y=30 446X-23 658,R2=0.999 5,结果表明花旗松素在1.4~117.0 μg/mL 线性关系良好。
2.13.7 精密度试验 取“2.13.3”项下含药肠灌流供试液适量,分别加K-R 试液稀释至质量浓度梯度为75、50、25 μg/mL,按“2.3.1”项下色谱条件1 d 内测定5 次,日内精密度RSD 分别为0.87%、0.09%、0.07%,表明日内精密度良好;连续测定5 d,日间精密度RSD 分别为为0.22%、0.10%、0.06%,表明日间精密度良好。
2.13.8 稳定性试验 取“2.13.3”项下含药肠灌流供试液适量,分别在室温下放置0、2、4、6、12、24 h,按“2.3.1”项下色谱条件进样测定,RSD 为1.25%表明稳定性良好。
2.13.9 重复性试验 取“2.13.3”项下含药肠灌流供试液适量6 份,按“2.3.1”项下色谱条件进样分析,结果花旗松素质量浓度的RSD 为0.97%,表明该试验重复性良好。
2.13.10 加样回收率试验 取“2.13.3”项下含药肠灌流供试液适量,分别加入低、中、高不同质量浓度的花旗松素对照品溶液,得到含药质量浓度梯度为60、50、40 μg/mL 的回收率样品溶液,按“2.3.1”项下色谱条件进样测定,计算加样回收率,花旗松素的加样回收率在98%~102%,平均加样回收率为100.11%,RSD 为1.50%,结果表明该方法符合检测要求。
2.13.11 大鼠在体单向肠灌流实验 选取健康雄性SD 大鼠60 只,随机分配各20 只至花旗松素组、Tax-PC 组、Tax-PC/BSA NPs 组,每组根据肠段再次随机分至十二指肠、空肠、回肠、结肠各5 只。实验前12 h 大鼠禁食不禁水,采用20%乌拉坦腹腔注射麻醉大鼠(注射剂量7 mL/kg),置于37 ℃恒温加热垫上固定。沿腹中线在腹腔中下部剪开3 cm的切口,分离目标肠段(十二指肠、空肠、回肠、结肠),在肠段的上下两端分别切开一个足以插入0.3 cm 直径玻璃管的小口,并使用灭菌手术线结扎固定。通过导管注入生理盐水(37 ℃预热)以清理肠段内容物,直至流出液无肉眼可见异物(图10)。将浸有生理盐水的脱脂棉覆盖在切断伤口处,保持肠道湿润、避免腹膜中液体及热量损失。肠段上端导管连接恒流泵,为平衡肠段,以0.2 mL/min 恒速灌流K-R 试液15 min,随后更换为花旗松素、Tax-PC、Tax-PC/BSA NPs 肠灌流液持续灌流肠段1 h,收集流出液。采用重量法对流出液体积进行校正。实验结束后,剪取经灌流的肠段,测量其长度与内径[17]。
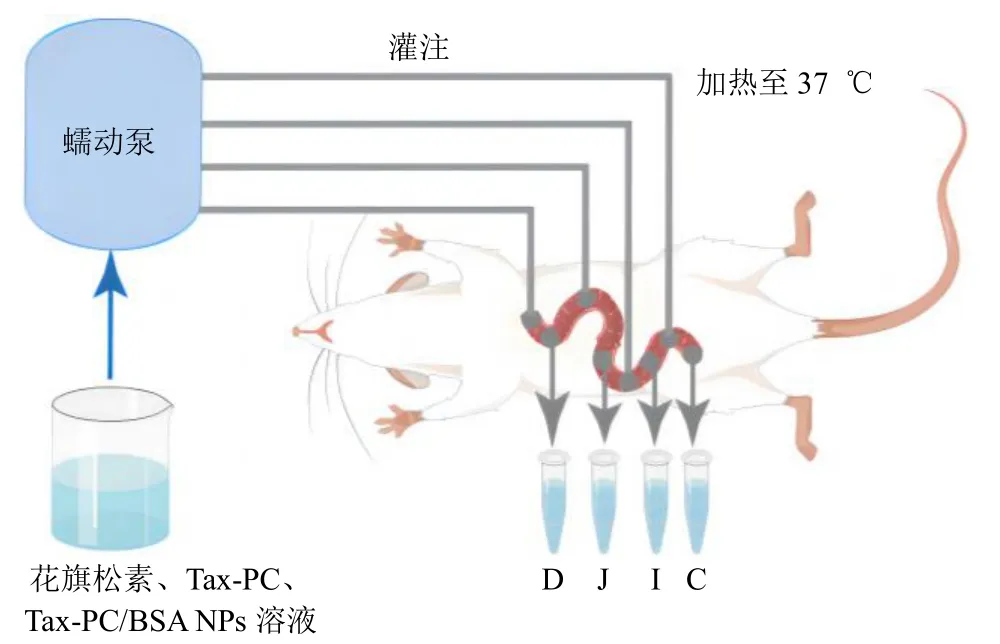
图10 大鼠在体单向肠灌流模型Fig. 10 In vivo unilateral intestinal perfusion model of rats
2.13.12 含药肠灌流液稳定性 取“2.13.3”项下含药肠灌流液,在37 ℃水浴条件下孵育0、30、60、90、120 min,各时间点取样1 mL,进样测定,将0 min 时药物质量浓度视为100%,计算孵育后花旗松素药物剩余率(即孵育前后供试溶液中花旗松素含量百分比)。结果,花旗松素、Tax-PC、Tax-PC/BSA NPs 灌流液剩余药量分别为(99.79±1.96)%、(100.48±1.42)%、(98.41±1.50)%,RSD 分别为0.97%、0.46%、1.05%,表明含药灌流液在K-R 试液中稳定性良好。
2.13.13 含药肠灌流液肠壁物理吸附 剪取肠段10 cm,生理盐水灌流冲洗,将黏膜层翻出,置于花旗松素、Tax-PC、Tax-PC/BSA NPs 含药肠灌流液中,在(37.0±0.5)℃水浴中恒温孵育,分别在0、2 h取样,进样测定,计算剩余率。结果表明,2 h 内花旗松素、Tax-PC、Tax-PC/BSA NPs 灌流液的剩余药量分别为(100.90±0.60)%、(101.24±0.75)%、(99.85±0.98)%,RSD 分别为0.43%、0.33%、0.62%,表明肠壁对灌流液中药物基本无吸附作用,对吸收测定无影响。
2.13.14 各肠段吸收情况 分别取花旗松素、Tax-PC、Tax-PC/BSA NPs 含药肠灌流液按照“2.13.6”项下方法操作,考察花旗松素、Tax-PC、Tax-PC/BSA NPs 在十二指肠、空肠、回肠和结肠段的吸收状况。按公式计算药物吸收速率常数(Ka)和药物表观吸收系数(Papp)。结果显示,花旗松素、Tax-PC、Tax-PC/BSANPs 在整个肠段均有吸收,且在十二指肠吸收最好,空肠次之,表明花旗松素主要吸收部位为肠道上部。形成Tax-PC、Tax-PC/BSA NPs 后花旗松素在各肠段的吸收均得到显著增强(P<0.05、0.01)。结果见表8。
表8 花旗松素及其不同制剂在不同肠段的Ka 和Papp (±s, n = 5)Table 8 Absorption rate constants of taxifolin and its different preparations in different intestinal segments Ka and Papp (±s,n = 5)

表8 花旗松素及其不同制剂在不同肠段的Ka 和Papp (±s, n = 5)Table 8 Absorption rate constants of taxifolin and its different preparations in different intestinal segments Ka and Papp (±s,n = 5)
与花旗松素组比较:*P<0.05**P<0.01;与Tax-PC 组比较:#P<0.05##P<0.01*P < 0.05**P < 0.01 vs taxifolin group;#P < 0.05##P < 0.01 vs Tax-PC group
肠段 Ka/(×10−3 min) Papp/(×10−3 cm∙min−1)花旗松素 Tax-PC Tax-PC/BSANPs 花旗松素 Tax-PC Tax-PC/BSANPs十二指肠 68.1±4.5 120.9±7.5** 137.6±2.2**## 8.9±0.8 23.0±3.4** 31.9±1.7**##空肠 60.2±4.5 114.8±8.5** 135.8±2.1**## 7.6±0.7 20.6±3.0** 30.6±1.4**##回肠 55.8±2.1 90.2±3.5** 131.3±3.1**## 6.9±0.3 13.3±0.8** 27.9±1.7**##结肠 28.1±7.2 37.6±2.0* 50.9±4.5**# 4.2±1.2 5.9±0.4* 8.9±1.2**#
Cin、Cout为进、出灌流液的质量浓度,Vin、Vout为进、出灌流液的体积,Q为灌流体积流量,r和L分别为肠段的半径及长度
3 讨论
课题组前期考察了多种制备方法将Tax-PC 载入白蛋白纳米粒中,包括去溶剂化法、纳米沉淀法、热凝胶法、喷雾干燥法、自组装法、NabTM技术等。综合多种制备方法,NabTM技术展现出较为突出的优势,制备得到的纳米粒粒径小、无表面活性剂参与,同时规避高温对脂质类制剂的氧化影响、保护蛋白性质等。NabTM技术利用了白蛋白的结构特点,在高剪切力下产生了气穴空化效应-超氧化物离子,通过氧化巯基残基或断裂二硫键,围绕花旗松素交联搭建新的二硫键,形成聚合物壳体[18]。目前,NabTM技术在白蛋白纳米粒的制备当中已获得广泛认可。
口服给药是最常用的临床给药方式,药物经口服后通常在小肠部位吸收,透过肠壁参与血液循环,进而发挥药物疗效。磷脂与药物结合,能够提高药物通过富含脂质的生物膜的能力,Tax-PC 明显提高了花旗松素的油水分配情况,而经过白蛋白纳米粒进一步包封后,其lgP值与磷脂复合物基本持平,仍适宜于药物吸收(lgP>1),侧面证明磷脂的两亲性在Tax-PC/BSA NPs 形成后得到保留,白蛋白纳米粒包封磷脂复合物,磷脂的2 条长脂肪链不参与该过程,仍然能够自由转动,形成亲脂外观。Tax-PC/BSA NPs 在Tax-PC 的基础上,进一步提高了花旗松素的平衡溶解度,改善药物口服吸收。
体外模拟消化释放过程中,Tax-PC/BSA NPs 的胃部累积释放率明显低于花旗松素,造成这种差异的原因可能是花旗松素经磷脂、牛血清白蛋白双层载体包封,在一定程度上维持了纳米粒结构的稳定性,经胃蛋白酶的消化作用后,部分BSA 需逐步分解,花旗松素才能缓慢释放。因此在胃部消化阶段,Tax-PC/BSA NPs 能够减缓花旗松素的释放,利于肠道对药物的吸收和利用。而在肠道消化环境模拟消化时,Tax-PC/BSA NPs 吸收状况明显优于花旗松素、Tax-PC,BSA 需经胰蛋白酶水解,胆盐辅助氧化分解,使纳米粒的结构破坏,药物快速释放,而磷脂等脂类物质的存在,也能够加速肠道消化。因此认为Tax-PC/BSA NPs 在一定程度上减少了药物在胃部吸收并增强了药物肠道定位释放,有效提高花旗松素的吸收状况。
基于此,本研究采用大鼠在体单向肠灌流模型,以更好地模拟体内肠道环境,比较不同剂型对花旗松素肠道吸收的改善状况,阐明其在各肠段吸收特征。结果表明,花旗松素在大鼠各肠段均有吸收,制备成磷脂复合物白蛋白纳米粒后,药物在各肠段的吸收均有显著提高(P<0.05、0.01),Tax-PC/BSA NPs 相较花旗松素,Ka在十二指肠、空肠、回肠、结肠分别提高2.02、2.26、2.35、1.81 倍,Papp分别提高3.58、4.03、4.04、2.12 倍,说明Tax-PC/BSA NPs 能够改善药物在体肠吸收状况,可为花旗松素新剂型的研发和临床合理应用提供新的选择。
此外,本课题组在后续研究中会进一步考察大鼠ig Tax-PC/BSA NPs 后的生物利用度,以验证制剂的合理性。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
临床小儿外科杂志(2021年10期)2021-11-05
家庭百事通·健康一点通(2019年8期)2019-08-29
中国继续医学教育(2018年35期)2018-12-26
现代营销(创富信息版)(2018年7期)2018-09-05
现代营销(创富信息版)(2018年2期)2018-02-10
澳门月刊(2017年12期)2017-12-15
中国兽医杂志(2016年7期)2016-08-30
现代营销(创富信息版)(2016年11期)2016-08-22
海南医学(2016年8期)2016-06-08
中成药(2014年11期)2014-02-28
