
机器学习在蛋白质疏水相互作用模型研究中的应用
2023-12-17 13:13:54冯晨博马维强
南京大学学报(自然科学版) 2023年6期
冯晨博,马维强,程 润,王 骏
(南京大学物理学院,南京,210093)
蛋白质是地球生命体系中重要的功能高分子,在新陈代谢、物质运输、形成细胞骨架、催化反应、免疫反应等过程中都发挥着积极的作用.在蛋白质结构形成过程和功能运动中,分子内相互作用是决定这些行为的核心物理要素,而在各种分子内相互作用中,疏水相互作用有十分重要的作用和意义.据估计,在单域球蛋白中,疏水相互作用占总体折叠自由能的70%~80%,这一现象启发了多种简化模型,如HP 模型等,这些模型为蛋白质折叠物理理论的研究提供了基本物理基础和典型物理模型.然而,疏水相互作用不同于经典相互作用等微观相互作用,它来源于蛋白质分子与溶剂水分子的相互作用、水分子之间的相互作用以及水分子部分的熵效应,要把这些效应唯象定义为蛋白质自身自由度的函数,常用的方法是通过对所有溶剂坐标积分来定义平均力势.但事实上,在微观时间和空间尺度上,还存在来自动力学的更多复杂性.在这种情况下,蛋白质体系中的疏水相互作用一定是蛋白质原子坐标的复杂多体函数,这使得描述疏水相互作用具有内秉的复杂性,成为一件非平凡的工作,定量刻画蛋白质-溶液之间相互影响的自由能函数一直是蛋白质物理研究的重要内容[1-2],常见的近似模型包括连续化溶剂(Continuum Solvent)模型[3]和原子溶剂化参数方法[4](Atomic Solvation Parameters Approach).前者用均匀可极化的连续介质代替添加进系统中的水分子来降低计算耗时,后者假设原子的自由能与其暴露在溶剂中的表面积成正比,将蛋白质的自由能写为:
其中,系数σi与原子种类有关,可以根据实验结果拟合得到.Ai为每个原子的溶剂可及表面积(Solvent-Accessible Surface Area,SASA)[5],具体是指探针溶剂分子滚过待计算分子的范德华表面时,其球心可到达的表面,等效于将待计算分子中的每个原子半径扩大一个探针分子半径后构成的范德华表面,而探针的大小反映了溶剂的微观结构特征.目前,研究溶剂可及表面积已成为分析蛋白质体系疏水相互作用的典型手段,在蛋白质与水的相互作用中,探针水分子半径通常取1.4 Å.目前已有多种相关手段可以计算SASA,但这些方法在计算成本和精确度方面仍难以平衡,相比于力场中其他种类的相互作用,一定程度上限制了蛋白质模型的简化.因此,找到高精度和高效率的疏水相互作用(或SASA)的计算方法受到了蛋白质模型研究的广泛关注.
近年来,人工智能技术发展迅猛,其不仅在图像/视频识别[6-9]、自然语言分析[10-13]、游戏博弈[14-17]等经典领域发挥十分重要的作用,还在疾病诊断[18-19]、药物设计[20-21]、结构生物学[22-23]、材料计算[24-25]乃至高能物理[26-27]等众多领域也发挥了十分积极的作用.这归因于深度学习(深度神经网络)方法在因果推断、模型表示等方面的突出弹性,也为研究复杂多体关联提供新的手段和工具.本文尝试运用深度神经网络的方法,基于深度势能架构,以解析计算结果为标记,对SASA这一物理量进行学习,实现高精度的SASA 预测,相应的计算速度也显著高于解析计算.这一模型实现了对蛋白质体系多体相互作用的重构,给出了一种准确且高效的计算手段,为提升蛋白质模拟效率提供支撑.
1 方法
监督学习是一种机器学习范式,是从成对的样本和与其相关联的标签中尝试学习一个函数,将输入的特征向量映射为输出,从而基于任意的可能输入来推断问题的答案.对于蛋白质SASA的预测问题,本文借鉴了基于深度神经网络的分子动力学模拟方案——深度势能分子动力学(Deep Potential Molecular Dynamics,DPMD)[28].
1.1 模型为了降低问题的复杂度,将蛋白质的总SASA 拆分为单个原子的SASA 之和,而单原子SASA 的预测依赖于原子的自身性质和其近邻环境.对于每个待计算的中心原子,首先提取与其相交的所有近邻原子,并求出近邻原子与中心原子的相对坐标,保证系统的平移对称性.然后,将近邻原子按照与中心原子的距离排序,保证系统的置换对称性.最后,使用两个最近邻原子建立坐标轴的参考向量(如图1 所示,图中原子j和k为原子i的最近邻和次近邻原子,l为任意待旋转的近邻原子.为局域坐标系的单位向量),并通过旋转矩阵调整所有近邻原子的空间取向:
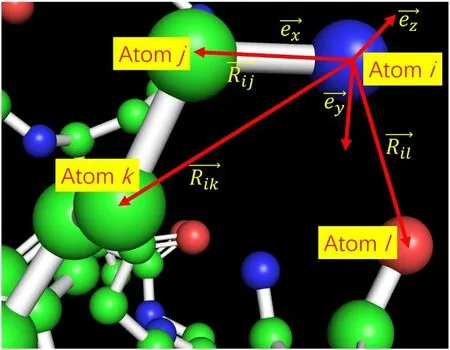
图1 原子i 的局域坐标系示意图Fig.1 The schematic diagram of the local coordinate related to the Atom i
其中,Rij=Ri-Rj,Rik=Ri-Rk,e[R]的作用是将R转化为单位向量.至此,具有平移、旋转和置换对称性的局域坐标系得以建立.在对原始坐标做整体平移、旋转或置换任意原子编号后,局域坐标系下的构型不会发生改变,对应了这些操作下不变的SASA.这压缩了原始数据的冗余,降低了势能面的维度.
最后,将旋转后的近邻坐标除以其与中心原子的距离,以分开输入角度和距离信息.据此,每个近邻原子与三条局域坐标轴夹角的余弦值、与中心原子的距离倒数以及自身半径,共计5 维向量作为近邻的描述符被输入网络.为了保证输入向量定长,只有离中心原子最近的固定个数的近邻信息得到保留.图2 展示了不超过一定近邻数的原子SASA 之和占全部原子SASA 的比例(SASA ratio)与近邻数M的关系.这一比例在近邻数小于40 时快速增长,近邻数为56 时对应比例已超过99%(如图中红色虚线所示),即包含更多近邻的原子SASA 之和不超过数据集中所有样本SASA 总和的1%,可以忽略,这反映了空间堆积的饱和性,此时即使选取更大的近邻数阈值,对SASA 的影响也很小.故选取截断近邻数Mcut=56,可以兼顾信息的完整性和效率.最后,为了便于神经网络的训练,所有输入数据都被标准化,即减去平均值后再除以标准差.

图2 一定近邻数以内的原子SASA 之和所占比例与近邻数M 的关系Fig.2 The ratio of SASA with an assigned number of neighbors
1.2 神经网络结构和训练过程得到的近邻信息被输入一个全连接的前馈网络,数据从输入层到输出层单向传播.该神经网络共有七个隐层,每层节点数分别为(1000,500,240,120,60,30,10),每层节点与下一层节点之间存在连接和偏置权重矩阵.数据经过权重矩阵的线性变换后,还会被施以非线性变换,以增强网络的非线性拟合能力.每层的非线性激活函数为Sigmoid.图3 直观展示了SASA 的预测流程和神经网络的架构.图3a 展示了完整的计算过程,Ri为蛋白质中原子坐标,N为蛋白质原子个数,{Rij}为原子i的近邻坐标集合,j为截断近邻数,{Dij}为原子i的环境的描述符集合,Si为原子i的暴露面积的预测值,SASA 为蛋白质总SASA 的预测值.图3b更加详细地展示了每个通道中Si的计算过程.
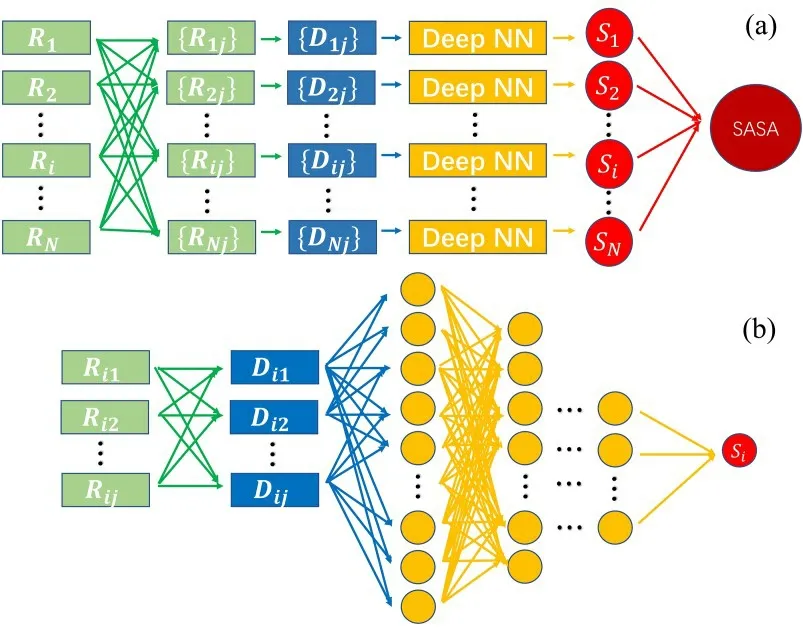
图3 SASA 预测方法和神经网络架构示意图Fig.3 Schametic diagram of SASA prediction workflow (a) and the related neural network architecture (b)
网络的损失函数由两部分组成:
losssingle为输出值与标签的均方误差(Mean-Square Error,MSE),losssum考察批次的整体误差.训练前期psingle占据主导,使权重从随机值快速拟合;训练后期psum逐渐占据主导,帮助稳定整体误差.只选用MSE作为损失函数会使网络的训练效果下降,且批次的整体误差会变得更不稳定.
训练过程中使用Adam 优化器调整权重,每次从数据集中随机抽取1000 个数据组成一个批次(batch)输入网络,训练100 个周期.每个周期调整权重的次数为训练集的样本数除以每个批次中的样本数,即N(train dataset)/N(batch),平均每个样本在每个周期输入网络一次.此处取N(batch)=1000,约为蛋白质包含的典型原子数.学习率的初始值为1×10-3,随训练周期衰减,前50 个周期内每周期衰减0.95 倍,后50 个周期调整为0.85 倍,最终值为2.68×10-8.为了缓解模型可能的过拟合,网络应用L2 权重正则化,为网络的损失函数添加了一项与权重值大小有关的惩罚:
使网络倾向于选择参数值分布的熵更小的简单模型,从而限制模型的复杂度.
1.3 数据集蛋白质数据来自SCOPe(Structural Classification of Proteins Extended)数据集[29-30],选用其中一个子集,其包含的蛋白质相互之间的序列相似度低于95%,既排除了相似度过高的同源序列,保障了相关数据的独立性,也避免数据集中出现过多的重复构型.SCOPe95 数据集共包含30201 个蛋白质,每个蛋白质包含的原子个数跨越三个数量级,大多数蛋白质的原子数约为1000 个.由于氢原子的体积过小,对SASA的影响可以忽略,所以在处理数据时所有氢原子都被去除.剩余原子共4120 万个,其中训练集、验证集和测试集分别占80%,10%和10%.
数据集的标签来源于ARVO 工具计算的解析值[1],使用相对简单的球极平面投影法[31].计算时每个原子半径都增加了1.4 Å,即一个水分子的半径.大约40%的原子暴露面积为0.图4 展示了单原子SASA 的分布情况.相应的统计信息:SASA 最小为0,最大为90.3 Å2,中位数为0.49 Å2,均值为6.95 Å2,标准差为12.5 Å2.因此,暴露面积的中位数仅为0.49 Å2.它们完全被埋在蛋白质内部,即使周围仍有空隙,水分子仍然无法与之接触.考虑到加入水分子半径后的单个原子总表面积约120 Å2,则蛋白质体系中约有6%的面积可与溶剂接触.
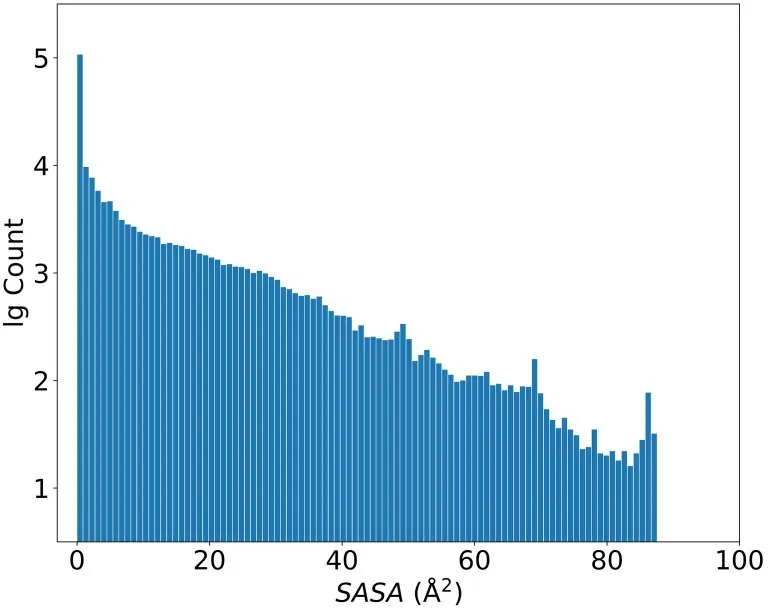
图4 蛋白质中20 万原子SASA 的直方图统计Fig.4 Histogram of atomic SASA in proteins
2 结果与讨论
2.1 模型训练使用Nvidia RTX 2070 训练约16 h 后得到的学习曲线如图5 所示,图中对应训练集的结果用红线表示,对应验证集的结果用蓝线表示,包括训练集和验证集的平均绝对误差(图5a)和整体误差losssum(图5b).测试集中,随着训练的进行,最终MAE和整体误差分别达到0.29 Å2和14.8 Å2.由于验证集的整体误差抖动幅度较大,图5b 应用了滑动平均,滑动窗口为10 个周期,验证集MAE下降到0.294 Å2,表明网络没有出现过拟合,且曲线与训练集MAE贴合紧密.对于整体误差,训练集呈平稳下降趋势,但验证集在前期偶尔出现强烈的反弹,在后期随着psum的增大逐渐趋于平稳,与训练集相差不大,收敛于14.2 Å2.结合训练集和验证集的结果可以证明,在尝试调整网络架构和超参数的过程中没有出现对验证集的过拟合,也说明整体误差对权重的变化比MAE更敏感.

图5 使用蛋白质全原子结构训练的模型的学习曲线Fig.5 The learning curve during the training processes with all-atomic protein structures
2.2 对SASA 的预测利用训练得到的神经网络对蛋白质SASA 进行预测.对单原子SASA 的预测结果如图6 所示,图6a 给出了单原子SASA预测值与相应解析值的比较(蓝色散点)和线性拟合(红线),图6b 为预测值的分段平均误差.可以发现,预测值与解析值的误差大致在0.5,没有随着标签值的增加而增加.

图6 (a)单原子SASA 的预测值和解析值对比;(b)单原子SASA 预测值的平均绝对误差与SASA 的关系Fig.6 (a) The comparison between the predicted and actual SASA for a single atom,(b) the MAE of predicted atomic SASA for the cases with various atomic SASA
选取2000 个蛋白质进行总SASA 的预测,其包含的原子全部属于测试集,结果如图7 所示.图7a 给出了预测值SASApre和相应解析值SASAact的比较(蓝色散点)和线性拟合(红线),其中,S0=1;图7b 为预测值与相应解析值的相对误差绝对值与体系原子数N之间的关系.结果表明,预测误差的绝对值在100以内的蛋白质超过97%,测试集蛋白质的总预测值比标签平均高出约7.5,约为一个原子的平均暴露面积,说明网络的预测存在约+0.08%的整体误差.对测试集误差取绝对值后除以各自标签,得到平均相对误差:且该相对误差δ与原子数N不存在相关性(如图7b 所示).为了说明本方法的精确度,选取三种计算SASA 的传统方法GetArea[32],FreeSASA[33]以及御茶水女子大学提供的计算网页[34],分别对33 个蛋白质进行测试.结果显示,三种方法对全部测试蛋白的计算值与ARVO 精确值的相对误差平均达到1.93%.因此,本方法与传统典型方法比较,误差缩小了近一个数量级.
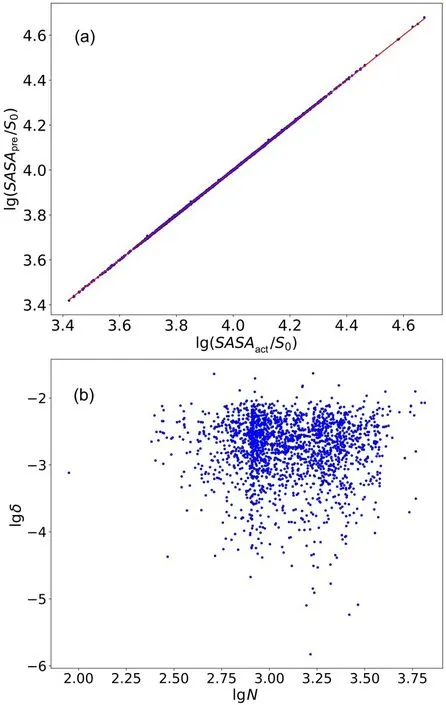
图7 (a)蛋白质总SASA 的预测值SASApre 和解析值SASAact 的对比;(b)预测相对误差δ 与原子数N 的关系Fig.7 (a) The comparison between the predicted and actual SASA for proteins,(b) the predicted relative error for the proteins with various numbers of atoms
2.3 对解折叠蛋白质的预测以上的预测蛋白质处于天然的折叠态,为了进一步验证网络对蛋白质SASA 的预测能力,将其应用到训练集中未曾出现的解折叠态蛋白质中,对测试集中被加热解折叠的三个蛋白质进行了测试,得到的结果如表1 所示.比较网络对这三个蛋白在折叠态和解折叠态的预测误差,可以看出,和折叠态相比,解折叠态的蛋白质SASA 都有升高,这符合折叠态是蛋白质自由能极小状态的观点.但其预测的相对误差都有显著增大,从0.5%扩大到1%左右,说明解折叠态中的原子环境与训练集中的折叠态存在较大差异,表明该网络对未曾接触的数据的可迁移性还有欠缺.

表1 折叠和解折叠态蛋白预测误差比较Table 1 The comparison between predicted errors for folded and unfolded proteins
2.4 网络精度及效率该网络的预测精度与训练集大小Nsample的关系如图8 所示,其中图8a 和图8b 分别刻画验证集的平均绝对误差MAE和整体误差losssum,图8c 为测试蛋白质体系的平均相对误差Dprotein,此时的训练集容量Nsample为1 万到3300 万.可以发现,预测精度与训练集容量的对数呈线性关系,训练集容量每增大一个数量级,MAE大约下降0.29 Å2.

图8 神经网络预测精度和训练集容量Nsample的关系Fig.8 The relations between the predicted accuracy and the size of training set
该神经网络的预测用时与使用C 语言的传统ARVO 工具相比,具有极大的优势.图9 对比了用两种方法计算不同原子数N的蛋白质所需的时间,其中,t0=1 s,本文提出的解析方法加速了约80 倍.两种方法的时间复杂度大致都为O(n),但神经网络方法将复杂度的系数降低了近两个数量级.以典型的蛋白质原子数1000 为例,神经网络预测用时约0.05 s.为了克服解释语言Python运算速度过慢的缺点,在建立本地坐标系时使用numba 中的JIT 解释器[35].
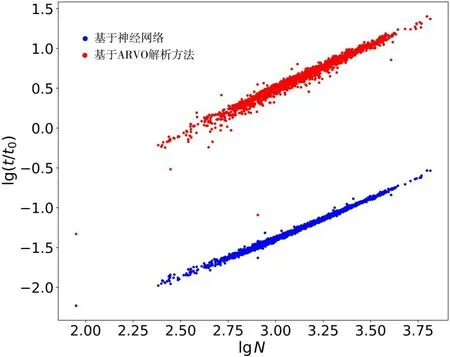
图9 不同方法对不同大小蛋白质SASA 的预测用时对比Fig.9 The time to calculate SASA for various sizes of proteins by different algorithms
2.5 拓展应用除了使用全原子的蛋白构型进行SASA 预测外,此方法也可应用到只有α 碳原子的蛋白质结构中.α 碳原子的SASA 指其所属残基所有原子SASA 之和SASAres.与全原子一样,同样存在大量α 碳原子的暴露面积为0,即其所属残基被完全包裹在蛋白质内部.将残基按亲疏水性质分类,亲水残基的平均SASAres达到70.9,而疏水残基平均只有35.4.亲水残基中被完全埋藏在内部的仅占2.5%,而疏水残基中这一比例达到12.2%,如图10 所示.图10a 和图10b 分别为亲水和疏水残基的情形,平均值分别为70.9和35.4.因此,蛋白质倾向于将疏水残基藏在内部,这也是其折叠的主要驱动力.

图10 亲水残基(a)和疏水残基(b) SASAres的直方图统计Fig.10 Histograms of SASA for the hydrophilic (a) and hydrophobic (b) residues
完整结构中的4120 万个原子包含520 万个α 碳原子,选取18范围内的α 碳近邻原子的信息输入网络,除了角度、距离和半径信息外,每个近邻的残基种类也通过字典编号并输入.经过嵌入层展开后,每个种类的索引被映射到20 维的向量中,并与角度、距离、半径信息连接,流入与全原子网络架构相同的全连接层.在前五个全连接层后分别插入一个Dropout 层,其在网络中引入噪声,有助于降低过拟合[36].典型蛋白质包含约100个残基,批次大小从全原子的1000 降为100.损失函数和学习率与全原子网络相同.520 万样本中,训练集占360 万,验证集和测试集各80 万.对2000 个蛋白质进行测试,得到平均相对误差为:
个别样本的误差δres超过10%.如图11 所示,图11a 为预测值和相应解析值的比较(蓝色散点)和线性拟合(红线),图11b 为预测值与相应解析值的相对误差绝对值与蛋白质链长Nres之间的关系.考虑到原子数与SASA 本就呈明显的线性关系,给网络仅提供α碳结构还是欠缺了较多的信息.

图11 (a)基于α 碳结构的蛋白质总SASA 预测值与解析值对比;(b)预测相对误差δres 与蛋白质链长的关系Fig.11 (a) The comparison between the predicted and actual SASA of proteins,(b) the predicted relative error for the proteins with various sizes
3 结论
深度学习的发展为研究多体相互作用带来了新范式,将神经网络方法应用到蛋白质SASA 的计算是一种新尝试.本文提取单个原子环境信息并转换到满足对称性要求的局域坐标系中,将数据集输入深度神经网络,通过最小化损失函数来优化网络权重参数,使其自行学习多体相互作用.将训练稳定的网络预测的结果与使用解析工具ARVO 计算的SCOPe 数据集中蛋白质所含原子的各自暴露面积进行比较,发现在对单原子SASA 预测时,MAE和整体误差分别为0.29和2.3%,对蛋白质SASA 预测的平均相对误差为0.26%.还分别利用解析方法和神经网络方法对随机选择的蛋白质SASA(单个蛋白质包含的原子数为100~10000 个)进行预测并记录所用的时间,发现和解析方法相比,在保证合理的准确度的前提下,本文提出的神经网络方法的预测速率提升近两个数量级.还对蛋白质的α 碳粗粒化结构进行了SASA 预测,精度比全原子下降了约一个量级,但与传统近似计算方法的精度相当,且仍能够保持较高的计算效率.因此,在利用机器学习对蛋白质全原子和粗粒化结构的SASA 预测中,未来可以构建更精巧的网络拓扑,更高效地提取原子构象的重叠关系.这也为动力学模拟提供了可能的支撑,结合更多真实的物理限制,使构象沿势能面的特定方向演化.
猜你喜欢
企业界(2024年8期)2024-07-05 10:59:04
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
今日农业(2021年19期)2022-01-12 06:16:32
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
环境保护与循环经济(2021年7期)2021-11-02 08:10:54
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
国外核新闻(2020年8期)2020-03-14 02:09:19
池州学院学报(2015年3期)2016-01-05 01:13:04
