
Unraveling the molecular mechanism of prion disease:Insights from α2 area mutations in human prion protein
2023-12-15 11:48RongriTan谈荣日KuiXia夏奎DamaoXun寻大毛WenjunZong宗文军andYoushengYu余幼胜
Chinese Physics B 2023年12期
Rongri Tan(谈荣日), Kui Xia(夏奎), Damao Xun(寻大毛),‡, Wenjun Zong(宗文军), and Yousheng Yu(余幼胜)
1Department of Physics,Jiangxi Science and Technology Normal University,Nanchang 330013,China
2School of Science,East China University of Technology,Nanchang 330013,China
Keywords: prion protein,mutations,misfolding,molecular dynamics simulations
1.Introduction
Transmissible spongiform encephalopathy (TSE), also known as prion disease, is a group of fatal degenerative diseases of the central nervous system that exist in many mammals due to prion isomerism.[1-3]At present,there is mad cow disease,scrapie in sheep,Creutzfeldt-Jakob disease(CJD)and fatal familial insomnia (FFI), Gerstmann’s syndrome (GSS),kuru disease and so on.[4,5]These diseases have been found to be associated with the conformational conversion of a normal cellular prion protein(PrPC)into a misfolded pathogenic scrapie form(PrPSc),and many factors can trigger the isomerization of PrPC to PrPSc.However,the conversion mechanism of PrPC to PrPSc and the pathological interaction of prions remain unclear.[6,7]
The natural state prion protein (PrP) has an N-terminal flexible region and a C-terminal globular folding structural domain.[8]The N-terminal of the mature prion protein has no specific structure, and the C-terminal globular domain contains mainly three orderedα-helix structural regions(α1,α2,α3), two very short anti-parallelβstrands (β1,β2), and a disulfide bond connectingα2 andα3.[9]It is now generally accepted that the C-terminal structural domain of PrPC begins to misfold by disrupting the region formed by the twoβstrands andα1 region and against the structural area ofα2-α3.It is shown that among these structures,there are 40 mutant sites in the PrP gene encoding,[10,11]which are mainly present in the C-terminal globular domain of PrP, especially in theβ2-α2 loop region and theα2-α3 interhelical interface.[12,13]These pathogenic mutations result in human diseases such as CJD,FFI, and GSS.For example, San Hadziet al.found that the pathogenic mutation H187R,which is closely related to GSS,can contribute to subdomain segregation and conformational transition of PrP by reducing the stability of the natural structure of prion protein.[14]
In recent years,a large number of experiments and computational simulations have reported that the key site of the conformational transition of prion proteins is in theα2 region, especially at theα2 C-terminal.[15-19]Mutation sites in this region play a crucial role in the misfolding of prion proteins.In hydrogen exchange combined mass spectrometry experiments, Laszloet al.found that stabilization of theα2 region prevents misfolding and aggregation of PrP.[15]In NMR studies,the presence of a chemical agent will make theα2 C-terminal show a great deal of structural flexibility.[20]In addition, many molecular dynamics simulations have also shown the important role of theα2 C-terminal in the conformational transition of prion proteins.Chakrounet al.carried out the molecular dynamics simulations on theα2-α3 region and found a stableβ-sheet structure in the protein structure.[21]A transition fromα-helix toβ-sheet was found at theα2 Cterminus during the conformational change.[16]These simulations also demonstrated that theα2 fragment plays an important role in prion misfolding and conformational transition.
To investigate the effects of mutations in theα2 region,molecular modeling techniques were employed to gain insights into the molecular mechanisms underlying the early induction of PrPC misfolding.In this work, two mutant sites,V180 and H187, were selected for analysis in theα2 region.Four systems were designed with these two mutant sites, including the wild-type (WT) structure, the V180I mutant(V180I),the H187R mutant(H187R)and the double mutations(Double).By comparing the properties of WT and several other mutations,such as hydrogen bonding,hydrophobic and electrostatic interactions, we aimed to understand the interactions between different systems.Our study analyzed the conformational fluctuations, hydrogen bonding, salt bridges and the changes in secondary structure during simulations.These mutations led to local changes in the natural structure of prion proteins.The purpose of this study was to explore the potential mechanism of prion diseases caused by mutations and provide valuable insights for future prion disease treatments.
2.Computational methods
In this work, the wild-type prion NMR structure (PDB ID: 1HJM)[22]was obtained from the Protein Data Bank(PDB)[23,24]with the pH of 7.0.The V180I and H187R mutations were altered by using the Pymol package to replace the corresponding residue of 1HJM.Four systems were studied here and their initial structures are shown in Fig.1, namely WT, V180I, H187R and Double, respectively.Each system was placed in a cubic box with a minimum distance of 10 ˚A between the protein and the box boundary, and the TIP3P water model[25]was used to fill the cubic box.Molecular dynamics simulations of all systems were performed using the GROMACS 5.1.5 package[26]with Amber99sb-ildn force field.Then,in order to make the simulated systems electrically neutral, sodium ions were added to the systems to neutralize each system.
For each simulation, the system underwent energy minimization using the steepest descent method in 1000 steps.It was then heated up from 0 to 300 K and equilibrated at 1 bar and 300 K.Equilibrium MD simulations of 1000 ps and production MD simulations of 500 ns were performed for each system in the NPT ensemble.During the minimization of equilibration, periodic boundary conditions were applied to thex,yandzdirections in the NPT ensemble.The SHARK algorithm was used for bond constraints in all simulations.The electrostatic interactions were handled by the particle mesh Ewald (PME) method[27]and the cutoff radius for Coulomb and van der Waals interactions were set to 10.0 ˚A and 14.0 ˚A,respectively.
To explore the mechanism that stabilizes the natural structure of PrPC,MD trajectories were analyzed from a structural and energetic perspective.After the simulation, we obtained the final conformation of the protein and placed it in Fig.S1.All trajectory analyses were performed in the GROMACS and VMD programs.[28]Root mean square deviation(RMSD)and root mean square fluctuation (RMSF) of the backbone atoms were monitored relative to the initial structure to assess conformational fluctuations and the stability of the MD trajectories.Then,the stability of the PrPC hydrophobic core was assessed by calculating the solvent accessible surface area(SASA)using VMD, and the radius of gyration (Rg) was used to measure the size variation of the protein.To examine inter-peptide interactions, inter-peptide backbone hydrogen bonds and salt bridges(SBs)were calculated.Furthermore, the evolution of secondary structure and the measurement of secondary structure percentage were determined throughout the simulation using the DSSP program.[29]
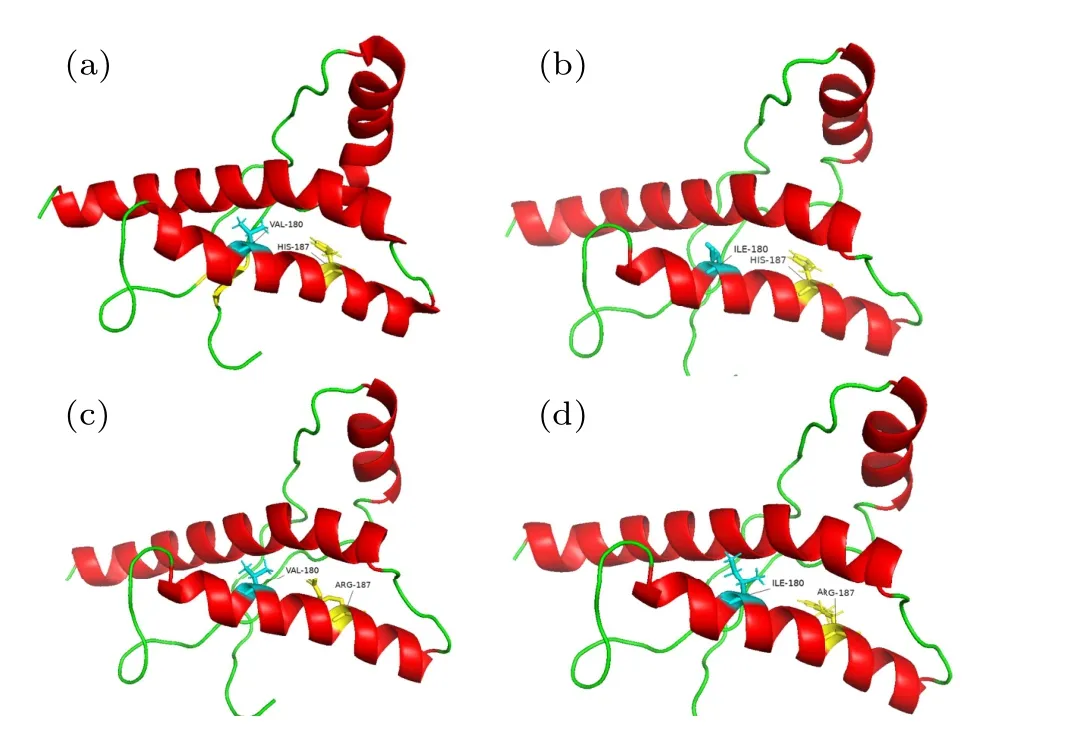
Fig.1.Initial structure of WT PrP and its variants.(a)WT PrP,(b)V180I variant,(c)H187R variant,and(d)Double variant.
3.Results and discussion
3.1.The stability of the α2 C-terminal and the hydrophobicity of the protein.
First, the RMSD of the Cαatom in the entire sequence was calculated to examine the structural changes caused by mutations in the globular structural domain.For each system,there are three parallel runs.Run 1 is shown in Fig.1, run 2 and run 3 are shown in Figs.S2 and S3, respectively.As shown in Fig.2(a), the RMSD values indicated that the WT reached equilibrium at 50 ns,whereas three mutants started to stabilize at 260 ns.It was also observed that the mutations exhibited higher structural fluctuation compared to the WT.The average RMSD values in the V180I,H187R,and Double systems were 0.26 nm,0.30 nm and 0.29 nm,respectively,while it is 0.23 nm in WT.Furthermore, the minimum deviation of the wild-type protein revealed that the wild-type conformation is the most stable,when compared to the three mutations.
To further explore the structural changes of each part,the RMSD value ofα1,α2, andα3 was calculated in each system.Figures 2(c)-2(e)show the volatility of each helix in different systems.It can be seen from the figure that each system was relatively stable in theα1 region.In contrast,for theα3 area,three mutants are more flexible.Interestingly,the mutant sites of the three mutations are all located in theα2 region.However, it has been found that the presence of a mutation at residue 187, such as H187R and Double, is more likely to causeα2 fluctuations in theα2 region.
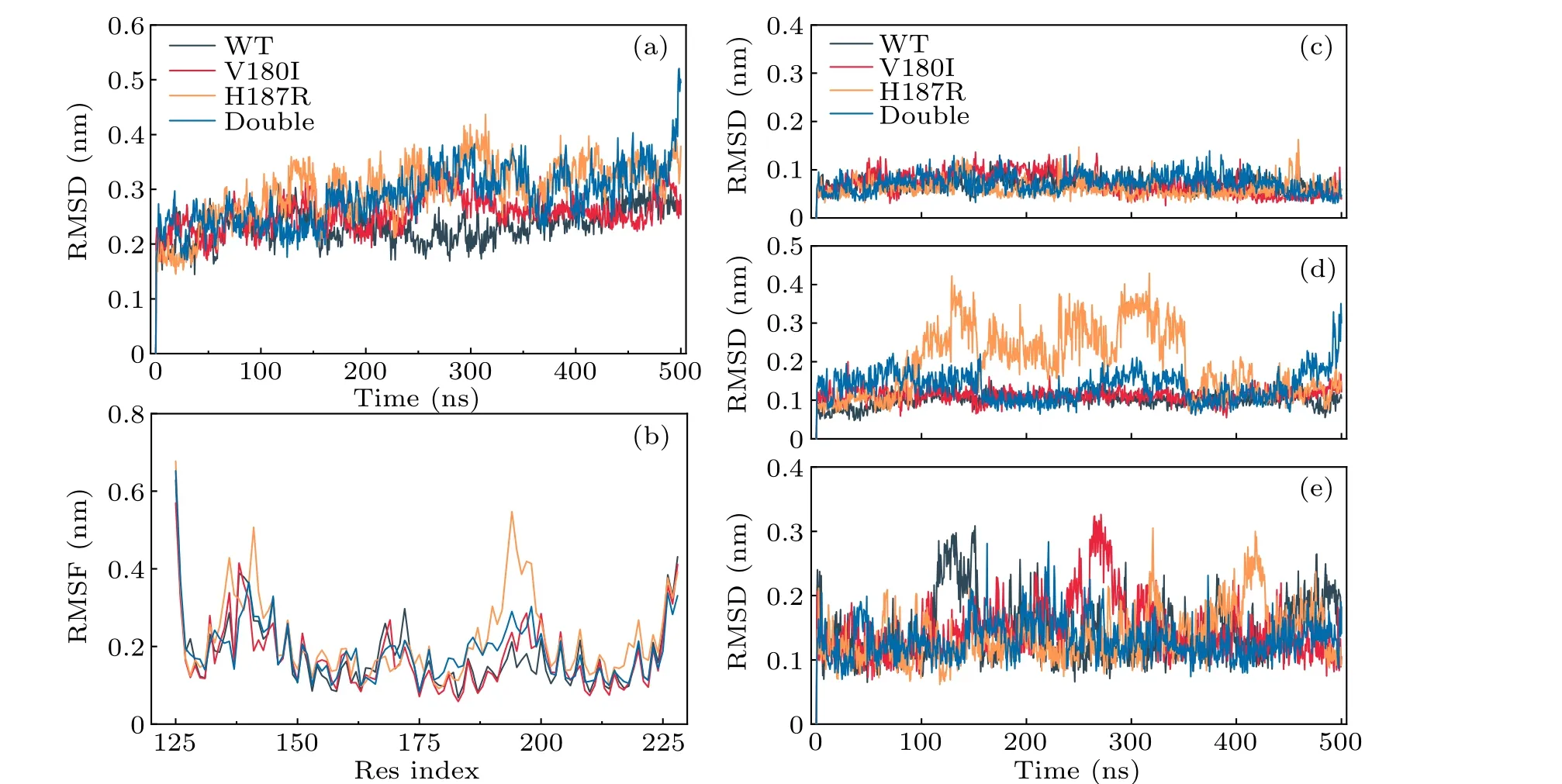
Fig.2.Stability and rigidity of WT PrP and its variants.(a) The RMSD values and (b) RMSF values of each system.(c)-(e) The backbone RMSD of three helixs(α1,α2,α3).
The RMSF serves as an important indicator of residue mobility around its average position, and can be used to analyze the dynamic stability of proteins.In Fig.2(b), the RMSF trends are shown to be similar in the four systems.In the H187R mutant, dynamic instability is observed in theβ1-α1 loop andα2-α3 loop, with maximum fluctuation of 0.50 nm and 0.54 nm.Conversely, the V180I mutant is dynamically stable for all regions of the protein.These mutations at residues 180 and 187 have led to a slight decrease in the flexibility of Double.Notably, all mutant systems have anomalous fluctuations near their mutation sites,especiallyα2 C-terminal.These fluctuations are considered as the first indication of conformational transitions.[11]Therefore, it can be inferred that all mutant systems undergo conformational fluctuations,and that point mutations may affect local protein interactions required for fiber species oligomerization.[30]
The time evolution of the SASA, hydrophobic SASA(Spho)and hydrophilic SASA(Sphi)is depicted in Fig.3.From the figure,we can find that theSphiis higher than theSphoin all systems.This phenomenon is due to the fact that hydrophobic residues and side chains of the protein can readily reside within the core, while the hydrophilic part is exposed to the solution.Furthermore, the difference betweenSphoandSphiis significantly increased in three mutant systems compared to the WT.Table 1 displays the specific solvent surface values that reveal that theSphoof WT is 3288 ˚A2,whereas theSphoof other three mutant forms have reduced to 3238 ˚A2, 3154 ˚A2,and 3184 ˚A2, respectively.In contrast, theSphihas increased to different degrees during the simulation.The transformation trends ofSphiandSphoare consistent with the study of the human prion protein.[31]It is well known that the unfolding of the protein releases hydrophobic residues,which increases the exposure hydrophobic surface.Furthermore,specific experimental results have demonstrated that protein denaturation is associated with an increase in hydrophobic surface exposure.[32,33]These results suggest that the onset of prion diseases may be related to prion protein denaturation caused by mutations.
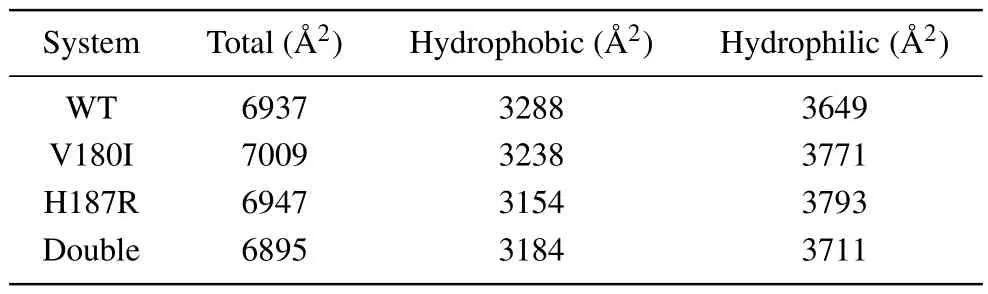
Table 1.The average of SASA values.
The Rg is a valuable parameter for measuring the effective size of a protein chain.Therefore, to comprehend the overall shape and size change in the mutant protein, we plot the function diagram of Rg over time and the probability distribution of Rg for Cαatom, as shown in Fig.4.Figure 4(a)reveals that the Rg value of WT is situated at the bottom of the depicted position,with the smallest Rg value.After 300 ns evolution, the Rg value of the Double is eventually stable at the highest Rg value of 1.49 nm.This shows that the overall size of the protein is altered by the mutation.In Fig.4(b),the V180I mutant form is observed to have the narrowest Rg distribution, indicating that V180I has the least conformational space.Both the double point mutation and H187R have a broader distribution than V180I, while the wild type has the broadest Rg distribution.It suggests that point mutations contribute to the folded conformation of the entire protein.
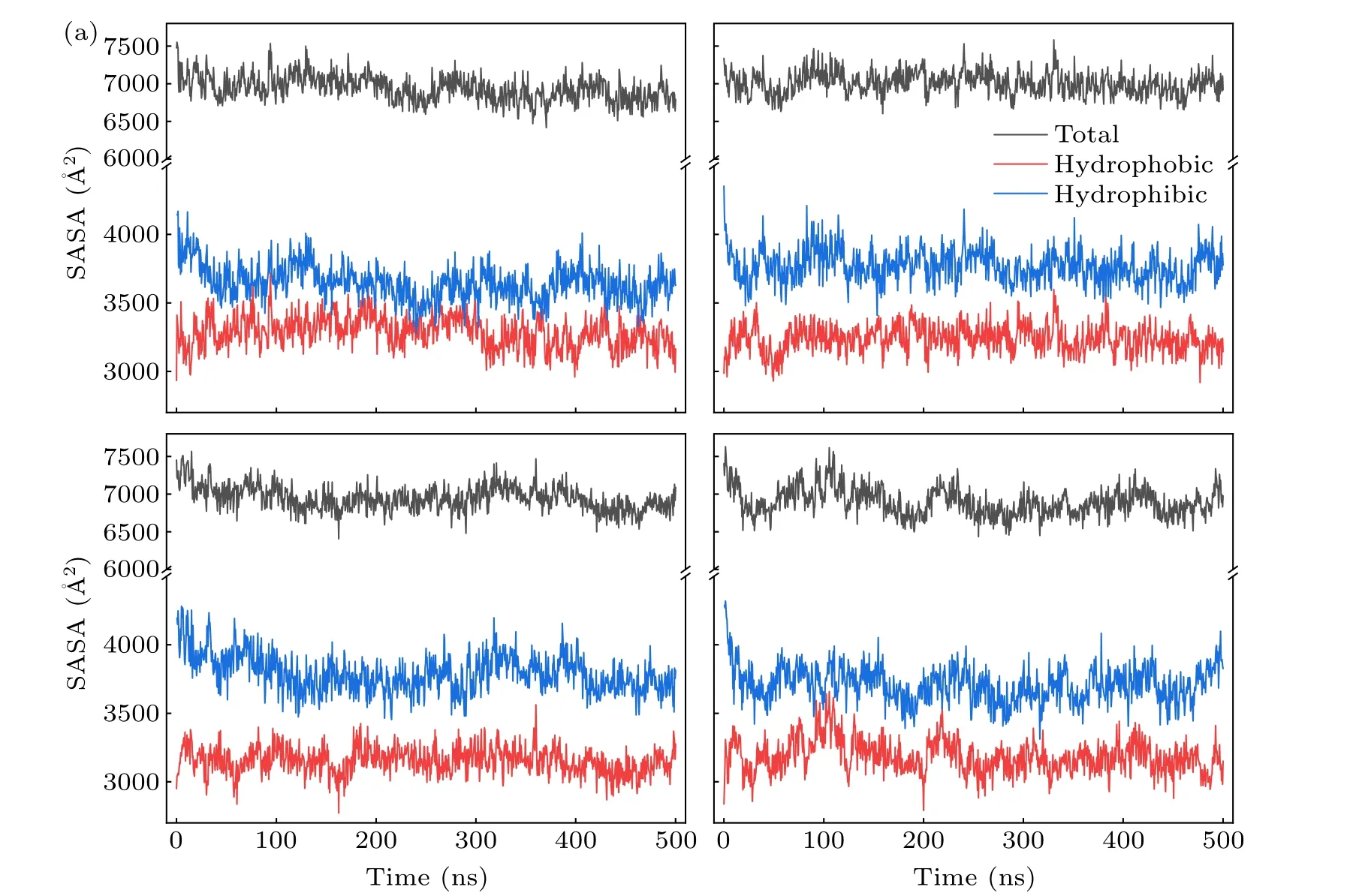
Fig.3.Time evolutions of the solvent accessible area of(a)WT,(b)V180I,(c)H187R and(d)Double.
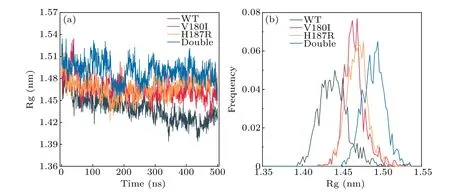
Fig.4.(a)The Rg value vs.time.(b)The probability distribution of radius of gyration of Cα.
3.2.Local disruption of hydrogen bonds and salt bridges
Some specific salt bridges can play a decisive role in stabilizing the folded structure of PrPC.[34]Even a small disruption of salt bridge accelerates the transition of the folded conformation toward an unstable state.[35]Therefore, the probability diagram of SBs of wild type and mutant are plotted in the Fig.5.For theα1 region, the SBs of V180I and H187R do not show significant changes.In the Double,the occupancy of D144R148 increased abnormally, resulting in a shortened distance between the two residues and verifying the result thatα1 has significant fluctuations in the Double.In theα2-α3 loop region,E196R156 was weakened in H187R and Double,which cause theα1 structural domain to move away from theα2-α3 core.This process led to the increased exposure of the hydrophobic core to the solvent environment.[36]Additionally,the salt bridges of D178R164 and D202R156 were partially lost in H187R and Double, causing the two substructural domains,β1-α1-β2 andα2-α3, to separate.For E207K204 and E211K204 in theα3 region,all three mutant systems experienced the total or partial loss, leading to instability in theα3 region.These mutations disrupted the native salt bridge network and potentially accelerated the conversion of PrPC to PrPSc,which may explain the occurrence of the disease.
To better understand the stability of each domain, the main chain hydrogen bonds of each domain are listed in Tables 2 and 3.The occupancy of hydrogen bonds over 75%is considered to be real.In Table 2, it can be observed that although the number of hydrogen bonds in theα1 region did not change for all four systems,the 144Asp-148Arg,which is found in all other systems,was not found in Double.And the distance of Double between 144Asp and 148Arg is shortened in salt bridge,which may also be the reason for the disruption of the hydrogen bonds.Moreover, the number of hydrogen bonds of V180I decreases significantly in theα2 region.Additionally, the mutant forms in theα3 region are deficient in the number of hydrogen bonds to some extent.These results indicate that the mutant forms have unstable structures in theα3 region, consistent with the finding of salt bridges.In Table 3, more hydrogen bond interactions are found in V180I betweenβ1 andβ2,as shown by the secondary structure content later,which is the reason for the increase ofβ-sheet.
In addition, it was shown that shortβ-sheet plays an important role in the conversion of natural prion proteins into pathogenic structures, with both elongation and depletion ofβ-sheet likely to be key processes.[37]129N:163O,129O:163N, 131N:161O, and 134N:159O, the four mainchain hydrogen bonds, are believed to provide supporting evidence for the presence of naturally occurringβ-sheet structures.[38]Figure 6(a) shows the occupancy of these four hydrogen bonds over time.The most significant change was observed in the hydrogen bond between 159O:134N, which showed an extremely weak appearance in V180I compared to the WT.The change may be due to the elongation of theβstructure (Table 2), causing disruption in the original hydrogen bond.On the other hand,in Double,where residue 180 is also replaced,the 159O:134N hydrogen bond is strengthened.This may be due to the mutation of residue 187 that stabilizes the presence of the hydrogen bond.However, for the rest of the hydrogen bonds in the four systems,no significant change in hydrogen bond occupancy was observed.
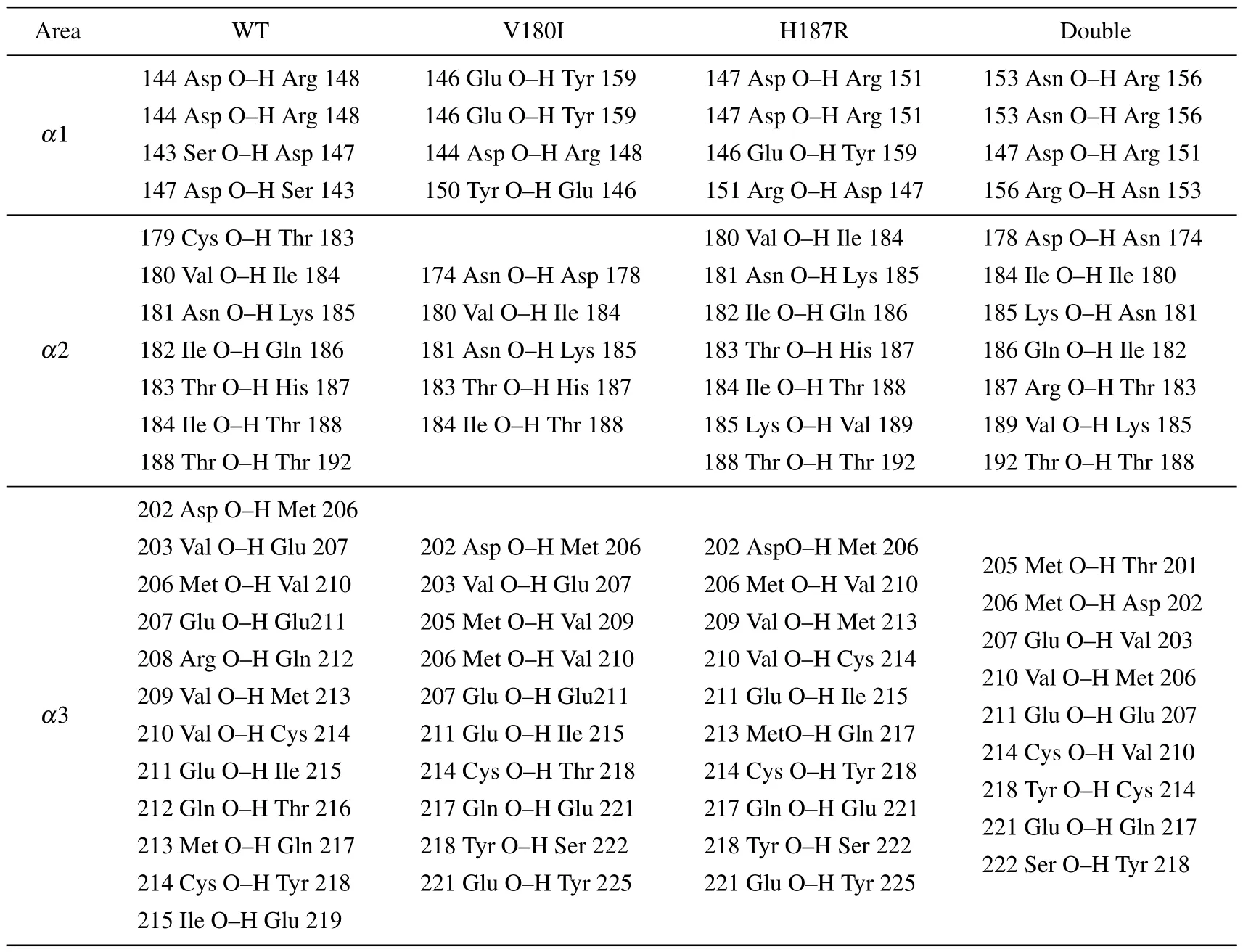
Table 2.The hydrogen bonds of helix areas.

Table 3.The hydrogen bonds of special sites.
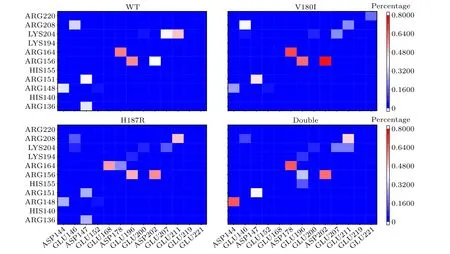
Fig.5.The salt-bridge heatmap of(a)WT,(b)V180I,(c)H187R and(d)Double.
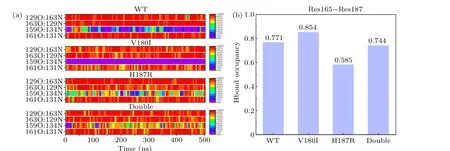
Fig.6.(a)Occupancy plots of different hydrogen bond pairs present in S1 and S2 strand of each system.(b)Occupancy plot of Res165-Res187 hydrogen bond.
The mutant protein undergoes some local conformational changes relative to the natural protein,especially near the mutation site.It can be seen from Fig.6(b) that the replacement of His187 with Arg187 results in the repulsion between Arg187 and Arg156 due to their positively charged side chains.This repulsion is influenced by strong electrostatic interactions.As a result,Arg187 experienced a significant deviation from Arg156, leading to an increase in the distance between these two residues.Figure 6(b) illustrates the bonding interaction of residues 165 and 187,whereby there is a significant decrease in H187R,indicating that Arg156 and Arg187 move apart from each other.This increased distance creates a gap in the protein region,thus exposing more protein sections to the solvent.Similarly,in the Double,the bonding between residue 165 and residue 187 is strengthened compared to H187R.This effect can be attributed to the mutation of residue 180,which facilitates the interaction between residues.In summary, it is not difficult to draw that Double has a slight inhibitory effect on the disruption of single point mutations (V180I and H187R).
3.3.The secondary structure of the protein
Here, in order to obtain detailed structural information of PrP in the four systems, secondary structure analysis is performed for all systems, and the results are shown in Figs.7 and 8.It can be observed from Fig.7 that WT and other mutations rarely change the natural structure of PrP,but they are easy to transform into disordered irregularly coil and turn at theα2 C-terminus.Previous studies have shown that the C-terminus ofα2 is highly capable of formingβ-sheet,and that stabilizing this region can prevent the misfolding and oligomerization of PrP.[39,40]Interestingly, Fig.7(b) reveals that V180I is unstable in theα2 region, causing it to extend intoβ-sheet formation.This observation highlights a negative correlation between the stability ofα22 and the propensity for structural misfolding.
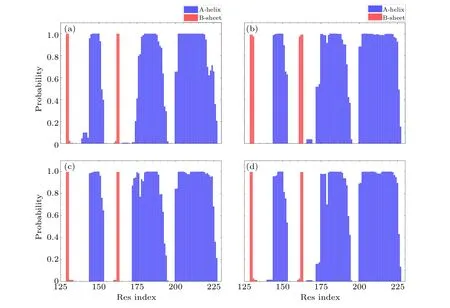
Fig.7.The probability of each residue for helix and sheet of(a)WT,(b)V180I,(c)H187R and(d)Double.
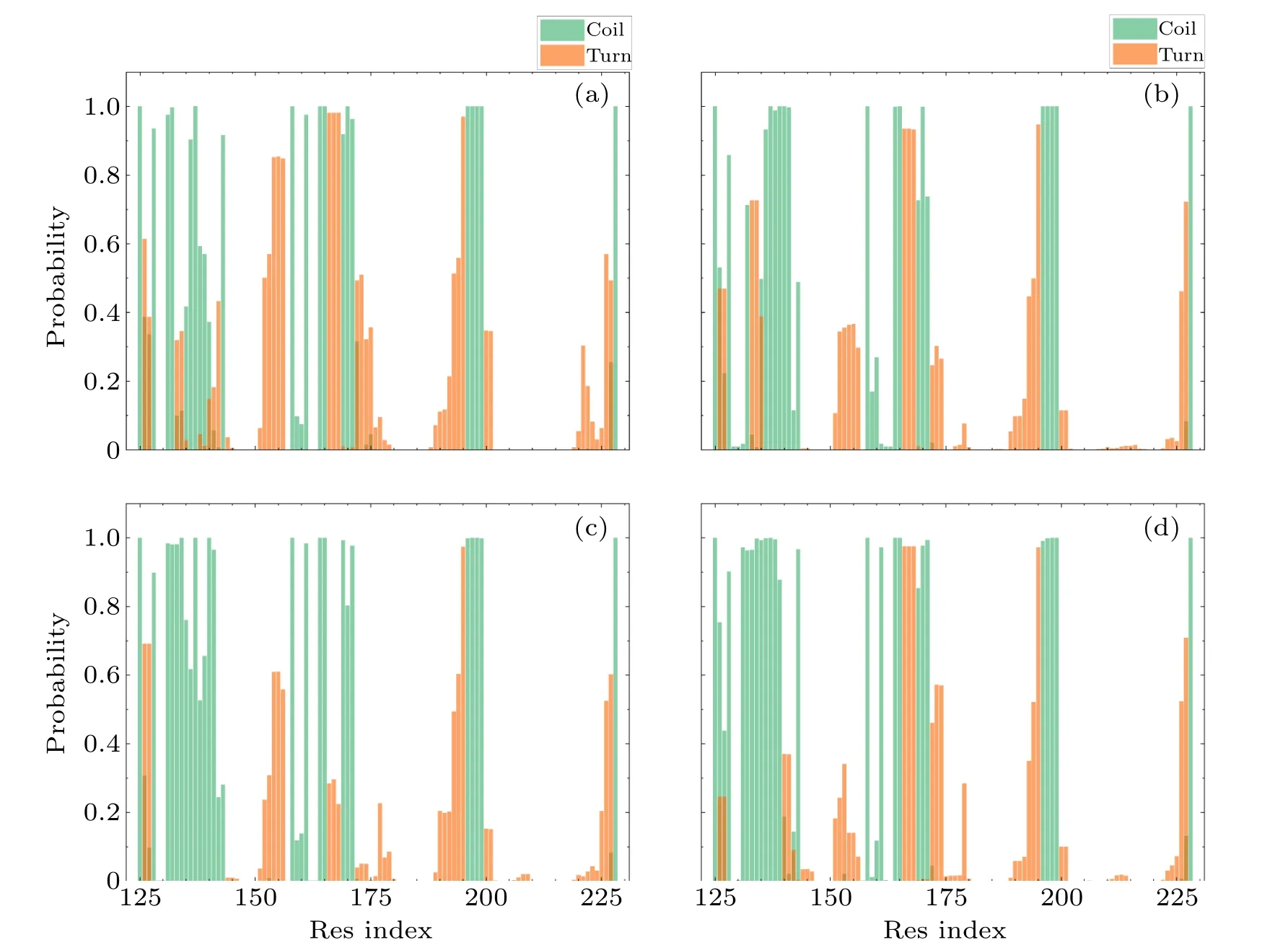
Fig.8.The probability of each residue for coil and turn of(a)WT,(b)V180I,(c)H187R and(d)Double.
In Fig.8,theβ1-α1 structural domains of all three mutations tend to form the irregular coil compared with theβ1-α1 region of the wild type.In Fig.9, we observed that the total content of random coils increased from 22%to 23%and 24%in H187R and Double,respectively.Furthermore,the content of turns in all helical regions decreased in all three mutant systems because partial turn structures were transformed into a helical structure due to the mutations,especially in theα3 region.In comparison to the WT, a significant increase in the content of theα-helix was observed in the three mutant systems.This observation can be seen in Fig.9.Meanwhile,the content of turn was reduced from 15%to 12%,9%,and 11%,respectively.This result suggested that mutations at theα2 C-terminus play a key role in the transformation of the natural structure, and it also demonstrated that theα2 C-terminus is unstable.
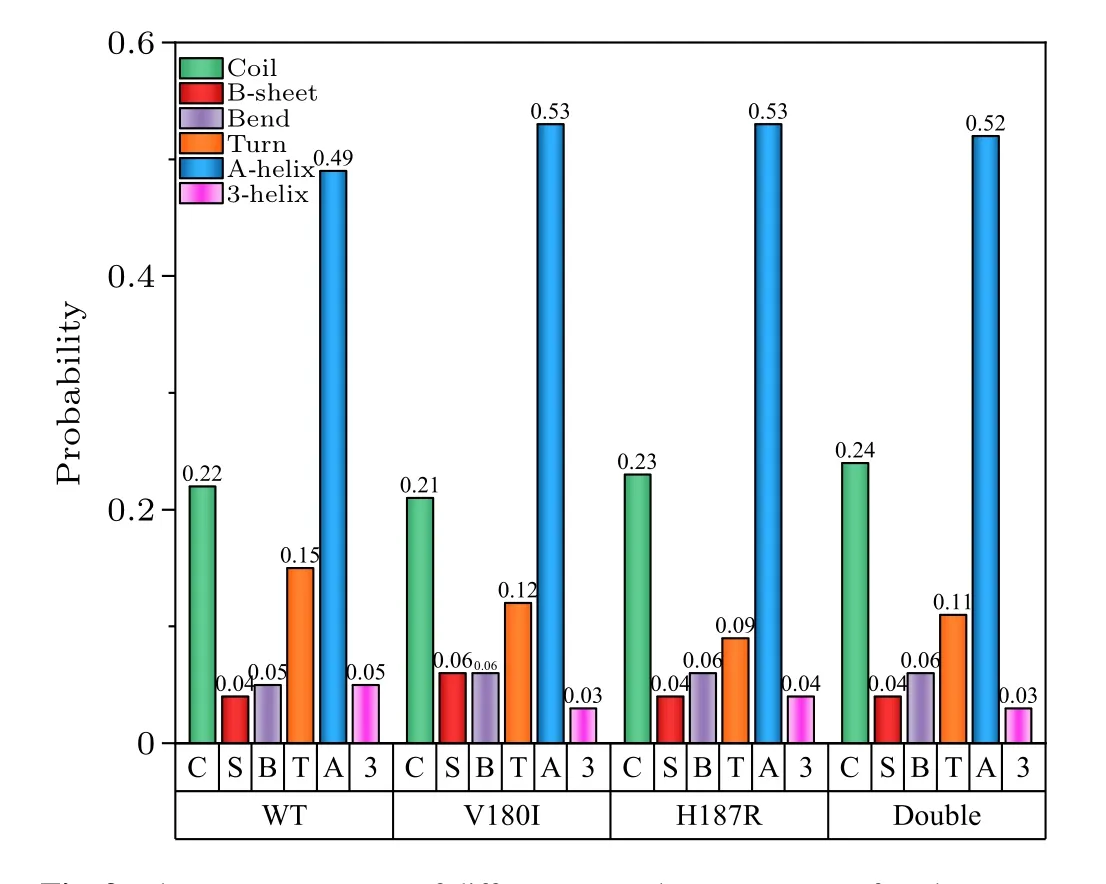
Fig.9.The average content of different secondary structures of each system.
4.Conclusion
The all-atom MD simulations were performed to investigate the effects and kinetic properties of mutations in theα2 region on the PrP structure,aiming to understand the molecular basis of disease-associated structural transitions.Although the differences between mutants and WT are usually limited,three mutations had distinct effects on the kinetics properties of PrP.Specifically, compared to other mutations, V180I resulted in the formation of an extendedβ-sheet in the secondary structure and an increased number of hydrogen bonds in the connection area ofβ1-β2.It also destabilized theα2 region,leading to a significant reduction in the number of hydrogen bonds onα2.In H187R and Double,key salt bridges such as E196R156 and D178R156 were weakened or disappeared,causing theα2 structural domain to become isolated from the others and exposing more residues to the solvent.The measurements of Rg and SASA indicated that several mutations induce minor conformational changes during the simulation.Therefore,we proposed that even small changes in natural proteins may be sufficient to trigger self-aggregation and cause diseases.
Data and software availability
The calculations and analysis of RMSD, RMSF,Rg, DSSP, salt bridges and Hbonds were performed with Gromacs software (https://www.gromacs.org/) version
5.1.5.The protein structure is available on Github(https://github.com/xk20/MMi-data-prion-validation/).
Acknowledgments
Project supported by the National Natural Science Foundation of China (Grant Nos.52073128, 12164002, and 11964012), the Foundation of Educational Committee of Jiangxi Province of China (Grant No.GJJ211112), and the Fund for Distinguished Young Scholars of Jiangxi Science & Technology Normal University (Grant No.2015QNBJRC002).

- Chinese Physics B的其它文章
- Diamond growth in a high temperature and high pressure Fe-Ni-C-Si system: Effect of synthesis pressure
- Multi-channel generation of vortex beams with controllable polarization states and orbital angular momentum
- Calibration of quantitative rescattering model for simulating vortex high-order harmonic generation driven by Laguerre-Gaussian beam with nonzero orbital angular momentum
- Materials and device engineering to achieve high-performance quantum dots light emitting diodes for display applications
- From breather solutions to lump solutions:A construction method for the Zakharov equation
- Complete population transfer between next-adjacent energy levels of a transmon qudit