
Canonical transient receptor potential channel 1 aggravates myocardial ischemia-and-reperfusion injury by upregulating reactive oxygen species
2023-12-14 10:54HuiNanZhanMenZhanWenTianWeiQuanFanSonShaoYuanLiuXiaoXiaoLiuDanMoYanSunYuanYuanGaoWenYeYinDaFenChanYanXinChenYeLeiZhouJinRuMenWeiCaoXiaoQianLi
Journal of Pharmaceutical Analysis 2023年11期
Hui-Nan Zhan ,Men Zhan ,Wen Tian ,Wei Quan ,Fan Son ,Shao-Yuan Liu ,Xiao-Xiao Liu ,Dan Mo ,Yan Sun ,Yuan-Yuan Gao ,Wen Ye ,Yin-Da Fen ,Chan-Yan Xin ,Chen Ye ,Lei Zhou ,Jin-Ru Men ,Wei Cao c,,**,Xiao-Qian Li ,*
a Department of Health Management, Second Affiliated Hospital, Fourth Military Medical University, Xi'an, 710038, China
b Department of Pharmacology, School of Pharmacy, Fourth Military Medical University, Xi'an, 710032, China
c Key Laboratory of Gastrointestinal Pharmacology of Chinese Materia Medica of the State Administration of Traditional Chinese Medicine, School of Pharmacy, Fourth Military Medical University, Xi'an, 710032, China
d Shaanxi Key Laboratory of “Qin Medicine” Research and Development, Shaanxi Administration of Traditional Chinese Medicine, Xi'an, 710032, China
e Institute of Materia Medica, School of Pharmacy, Fourth Military Medical University, Xi'an, 710032, China
fDepartment of Ultrasound Diagnostics, Second Affiliated Hospital, Fourth Military Medical University, Xi'an, 710038, China
g Department of Pharmacy, School of Chemistry & Pharmacy, Northwest A&F University, Yangling, Shaanxi, 712100, China
Keywords:TRPC1 Myocardial ischemia/reperfusion Reactive oxygen species OGDHL
ABSTRACT The canonical transient receptor potential channel(TRPC)proteins form Ca2+-permeable cation channels that are involved in various heart diseases.However,the roles of specific TRPC proteins in myocardial ischemia/reperfusion (I/R) injury remain poorly understood.We observed that TRPC1 and TRPC6 were highly expressed in the area at risk (AAR) in a coronary artery ligation induced I/R model.Trpc1-/-mice exhibited improved cardiac function,lower serum Troponin T and serum creatine kinase level,smaller infarct volume,less fibrotic scars,and fewer apoptotic cells after myocardial-I/R than wild-type or Trpc6-/- mice.Cardiomyocyte-specific knockdown of Trpc1 using adeno-associated virus 9 mitigated myocardial I/R injury.Furthermore, Trpc1 deficiency protected adult mouse ventricular myocytes(AMVMs) and HL-1 cells from death during hypoxia/reoxygenation (H/R) injury.RNA-sequencing-based transcriptome analysis revealed differential expression of genes related to reactive oxygen species(ROS)generation in Trpc1-/- cardiomyocytes.Among these genes,oxoglutarate dehydrogenase-like (Ogdhl)was markedly downregulated.Moreover,Trpc1 deficiency impaired the calcineurin(CaN)/nuclear factorkappa B(NF-κB)signaling pathway in AMVMs.Suppression of this pathway inhibited Ogdhl upregulation and ROS generation in HL-1 cells under H/R conditions.Chromatin immunoprecipitation assays confirmed NF-κB binding to the Ogdhl promoter.The cardioprotective effect of Trpc1 deficiency was canceled out by overexpression of NF-κB and Ogdhl in cardiomyocytes.In conclusion,our findings reveal that TRPC1 is upregulated in the AAR following myocardial I/R,leading to increased Ca2+ influx into associated cardiomyocytes.Subsequently,this upregulates Ogdhl expression through the CaN/NF-κB signaling pathway,ultimately exacerbating ROS production and aggravating myocardial I/R injury.
1.Introduction
Myocardial ischemia/reperfusion (I/R) injury is a pathophysiological process that occurs when blood flow to the myocardium is restored following a period of myocardial ischemia.It can lead to increased infarct size,tissue damage,and myocardial dysfunction[1].An increase in cytosolic Ca2+level plays a critical role in initiating cardiomyocyte disorders that occur in response to I/R [2,3].Although a set of Ca2+entry pathways,including the L-type Ca2+channel (LTCC) and Na+/Ca2+exchanger (NCX),have been implicated in mediating cardiomyocyte Ca2+mishandling,these contractile Ca2+proteins alone do not account for the activation of the signaling pathways that cause cardiac diseases[4,5].In addition to these contractile Ca2+proteins,cardiomyocytes are equipped with several Ca2+-handling proteins that may regulate signaling pathways or the Ca2+homeostasis of cellular compartments [6].Emerging evidence suggests that the Ca2+signaling is involved in the development of heart diseases such as cardiac hypertrophy and heart failure[5].However,the sources and cellular locations of the Ca2+ion involved in myocardial injury after I/R remain unclear.
The canonical transient receptor potential channel (TRPC)proteins,consisting of four subgroups (TRPC1,TRPC2,TRPC3/6/7,and TRPC4/5),form Ca2+-permeable cation channels that play a role in various heart diseases [7].While TRPC proteins are expressed at low levels in healthy adult cardiomyocytes,the expression and activity of certain isoforms can be upregulated in heart diseases [8-11].TRPC proteins are known as a family of cation-channel proteins that can generate complex spatiotemporal Ca2+patterns [12].TRPC channels can generate local Ca2+microdomains or modify the duration and amplitude of local Ca2+levels [13].TRPC channels have been regarded as initiators of the Ca2+-dependent signaling that leads to heart failure,hypertrophy,and pathological cardiac remodeling [14].Cardiac-specific overexpression of TRPC6 isoforms in transgenic mice stimulates Ca2+-dependent signaling pathways that mediate pathological cardiac hypertrophy and heart failure[15].Additionally,TRPC1 deficiency prevents the development of cardiac hypertrophy in mice subjected to pressure overload [16].Nevertheless,the role of TRPC proteins in myocardial I/R and the underlying mechanisms remain elusive.
In this study,we aimed to investigate the expression and functional significance of TRPC1 and TRPC6 in the area at risk(AAR) following myocardial I/R.We observed a significant upregulation and activation of TRPC1 in the myocardium of mice post-I/R and in cultured cardiomyocytes subjected to hypoxia/reoxygenation(H/R).TRPC1 upregulated oxoglutarate dehydrogenaselike (Ogdhl),which subsequently increased the production of reactive oxygen species (ROS),exacerbating I/R-induced cardiac injury.Furthermore,TRPC1 facilitated the upregulation ofOgdhlvia the Ca2+/calcineurin (CaN)/nuclear factor-kappa B (NF-κB)signaling pathway.These findings suggest that TRPC1 may represent a novel therapeutic target for the management of myocardial I/R injury.
2.Materials and methods
2.1.Animals
Trpc1-/-mice andTrpc6-/-mice on the 129Sv:C57BL/6J (1:1)background were generated as described before (Figs.S1 and S2)[17,18].These mice were back-crossed to C57BL/6J mice for more than 18 generations.For the in vivo experiments,8-week-old male mice were used alongside age-matched male C57BL/6J wild-type(WT) mice as controls.Mice were housed under a 12 h/12 h(light/dark) cycle and a constant temperature with ad libitum access to food and water.They were treated in compliance with the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences,China).All the animal experiments were approved by the Ethics Committee of First Affiliated Hospital of the Fourth Military Medical University (Approval No.: KY20194099).Euthanasia was confirmed via the failure of the animal to respond to a firm toe pinch.
2.2.In vivo adeno-associated virus serotype 9 (AAV9)-mediated cardiomyocyte-specific gene silencing or overexpression
Recombinant AAV9 was generated by harboring short hairpin RNAs(shRNA)-Trpc1or cloning theOgdhlorNF-κBcoding sequence into SalI and NheI restriction sites of the AAV9-cardiac troponin T(cTnT) receiving vector (Hanbio,Shanghai,China).To purify the AAV9 particles,they were first subjected to iodixanol step-gradient centrifugation,followed by fast protein liquid chromatography(GE Healthcare Life Sciences,Beijing,China),and then filtered through a 0.22-μm filter.Finally,the particles were titrated using quantitative polymerase chain reaction (q-PCR).To deliver the shRNA-carrying AAVs into the heart of mice,intra-myocardial injection was used as previously researched [19].Briefly,mice were anesthetized via 2%-isoflurane inhalation,and a skin incision was made on the chest.The pectoral muscle was incised.The heart was exposed through a hole at the 4th intercostal space.AAV9(1×1012μg/mg,45 μL)was slowly injected into the left-ventricle free wall.Afterward,the heart was replaced into the chest,the pneumothoraxes were evacuated,and then the skin and muscle incisions were sutured.After 21 days,the mice were subjected to myocardial I/R as described below.
2.3.Myocardial I/R injury
The I/R injury in mice was performed as in a previous report[20].Mice were anesthetized via 2%-isoflurane inhalation,the skin on the left chest was incised,and a hole was made at the 4th intercostal space.The pleural membrane was opened,and the heart was then carefully popped out.The left anterior descending(LAD)artery was located,and a slipknot was tied around the left coronary artery 2 mm from its origin by using a suture (6-0) and then ligated.The heart was then immediately placed back into the chest,and evacuation of the trapped air was performed manually to prevent pneumothorax.Muscle and skin were then sutured.In the sham group,a surgical procedure was performed that closely mirrored the experimental procedure,with the exception that the LAD was not ligated.Reperfusion was operated 30 min after ischemia by releasing the slipknot.
2.4.Primary culture of neonatal mouse ventricular myocytes
Neonatal mouse ventricular myocytes (NMVMs) were isolated and cultured as described previously [21].Briefly,the ventricles of WT,Trpc1-/-,andTrpc6-/-neonatal(1-day-old)mice were excised and then minced in Hank's balanced solution.They were then digested with 0.625 g/L collagenase II(Sigma-Aldrich,St.Louis,MO,USA) at 37°C.Subsequently,the digest was filtered and then centrifuged at 1,000gfor 10 min.Afterward,the pellet was resuspended in M-199 medium(Noninbio,Shanghai,China)with 5%(V/V)fetal bovine serum (FBS;Merck,St Louis,MO,USA) and 5 mM D-glucose and then incubated in a culture flask at 37°C for 40 min to remove non-cardiomyocytes.Then the cardiomyocytes were plated at a density of 1×106cells/mL in M-199 medium with 10%(V/V)FBS.A confluent monolayer of spontaneously beating cells was formed within 48 h.
2.5.Primary culture of adult mouse ventricular myocytes
Adult mouse ventricular myocytes (AMVMs) were isolated and cultured as previously described [22].Briefly,after the hearts of deeply anesthetized WT,Trpc1-/-,andTrpc6-/-adult mice were quickly excised,cannulated via the aorta,and then perfused at 37°C as follows.The hearts were first perfused for 3 min with a Ca2+-free bicarbonate-based buffer containing 120 mM NaCl,5.4 mM KCl,1.2 mM MgSO4,1.2 mM NaH2PO4,5.6 mM glucose,20 mM NaHCO3,10 mM 2,3-butanedione monoxime,and 5 mM taurine (Sigma-Aldrich) followed by a buffer containing 0.4 mg/mL collagenase type B (Roche,Basel,Switzerland),0.3 mg/mL collagenase type D(Roche),and 0.03 mg/mL protease type XIV (Sigma-Aldrich).
Afterward,the ventricles were separated from the atria and then minced as described above in Section 2.4.AMVMs were isolated by mechanically pulling them apart and then filtered using a sterile 250-μm filter top.The remaining pellet was resuspended in Tyrode's solution by a stepwise increase of the Ca2+concentration to 1.2 mM.Then,the cells were plated on cell-culture dishes precoated with laminin.After 4 h,round and unattached cells were removed.The basal culture medium was modified M-199 containing 2 mM carnitine,5 mM creatine,and 5 mM taurine.
2.6.H/R
H/R was performed in AMVMs as previously described [23].Briefly,AMVMs were subjected to 5 h of hypoxia followed by 1 h of reoxygenation.To simulate hypoxia,the culture medium was replaced with a solution of 13.6 mM NaCl,14.8 mM KCl,1.2 mM KH2PO4,1.2 mM MgCl2,1.8 mM CaCl2,and 3.4 mM NaHCO3,and AMVMs were subjected to hypoxia by incubating them in an oxygen-free atmosphere (95% N2and 5% CO2).Reoxygenation was conducted by placing the cells back in a normoxic incubator and replacing the hypoxia medium with the normal culture medium.For NMVMs,6 h of hypoxia followed by 9 h of reoxygenation was used.For HL-1 and HEK 293 cells,cells were cultured in sterile Dulbecco's Minimal Essential Medium (DMEM,Gibco,NY,USA)supplemented with 10% (V/V) phosphate-buffered saline (PBS)(Merck)and 1%penicillin-streptomycin(Sigma-Aldrich),and 6 h of hypoxia followed by 9 h of reoxygenation was performed.
2.7.Live/dead cell-viability assay
AMVMs were stained with 2 μM calcein acetoxymethyl (AM)(Life Technologies,Carlsbad,CA,USA) and 5 μM ethidium homodimer-1(Life Technologies)for 20 min at room temperature.Then they were examined using an Olympus IX71 fluorescence microscope (Olympus Corporation,Tokyo,Japan).Live and dead cells appear green and red,respectively.
2.8.Flow cytometric analysis
Apoptosis and necrosis rates were measured using flow cytometry with an Annexin V-fluorescein isothiocyanate/prodium Iodide (PI) detection kit (Biovision Inc.,Mountain View,CA,USA)[24].NMVMs or HL-1 cells subjected to H/R were collected and then stained with Annexin V and PI according to the manufacturer's instructions.Apoptosis and necrosis rates were determined using the Cell Quest software(BD Biosciences,Franklin Lakes,NJ,USA).
2.9.Measurement of mitochondrial membrane potential (ψm)
Mitochondrial ψm was determined via JC-1 staining [25].HL-1 cells subjected to H/R were washed with and then incubated with JC-1 (Beyotime Institute of Biotechnology,Nanjing,China)at 37°C for 20 min in the dark.Fluorescence intensity was measured first at an excitation/emission of 485/580 nm (red)and then at an excitation/emission of 485/530 nm (green) by using an inverted fluorescence microscope (Olympus Corporation).The red/green ratio was analyzed using the ImageJ software.
2.10.TRPC-mediated Ca2+ entry to cells
AMVMs or HL-1 cells were incubated with 5 μM Fura-2/AM(Molecular Probes,Eugene,OR,USA) for 30 min at 37°C.Subsequently,the cells were perfused with a Ca2+-free normal Tyrode's solution,and then 5 μM cyclopiazonic acid(CPA,Sigma-Aldrich) and 10 μM oleoyl-2-acetyl-sn-glycerol (OAG,Sigma-Aldrich) were added for 10 min to activate TRPC channels.Measurements were performed using a monochromator (Polychrome V,TILL-Photonics,Munich,Germany)-equipped inverted microscope with a Fluar objective.Every 1.2 s,Fura-2 was alternately excited at 340 nm and 380 nm for 30 ms each,and the emitting fluorescence intensity (F340 and F380,>510 nm) was recorded using a cooled charge-coupled device camera.Ratio images were calculated from the F340 and F380 pictures after background correction.Single cells were marked as regions of interest,and F340/F380 versus time was plotted.The cells were then provided with a buffer containing 1.8 mM Ca2+to assay for TRPC-mediated Ca2+entry.In some experiments,the LTCC antagonist nifedipine (10 μM;Sigma-Aldrich) or TRPC antagonist SKF96365 (5 μM;Sigma-Aldrich) was added 30 min before the treatment of the cells with CPA and OAG.
2.11.RNA sequencing (RNA-seq) analysis
The hearts of WT,Trpc1-/-,andTrpc6-/-mice subjected to myocardial I/R were harvested 24 h after reperfusion,and the blood was washed out with ice-cold PBS.The right ventricle and atria were trimmed away.Then,the AAR was dissected from the left ventricle.Total RNA was extracted from the AAR.RNA-seq was performed using an Illumina platform(Los Angeles,CA,USA).Briefly,the RNA samples were sequenced in an Illumina HiSeq 4000 instrument by using 150 cycles.Image analysis and base-calling were performed by Solexa pipeline V1.8(Off-Line Base Caller software,version 1.8).The reads were trimmed using pass FastQC version 0.11.5 filter (FastQ Screen,Cambridge,UK) to remove low-quality reads and then applied to the mouse reference genome (GenCode mm10) and mouse transcriptome (GenCode mm10) by using Hisat2 (version 2.0.5,Amazon,Annapolis,MD,USA).Transcriptional abundance was estimated using Stringtie (version 1.3.1c;Amazon).The genetranscription levels and major changes in transcription levels were calculated using Ballgown (version 2.8.4;Amazon).Differentially expressed genes (DEGs) in Gene Ontology (GO) terms or signaling pathways were identified via GO analysis.Three replicates were independently performed.DEGs with a fold change >|1.5|andP<0.05 were considered to be statistically significant.
2.12.Echocardiographic measurements
Left ventricular function was analyzed using echocardiography 24 h post-I/R.Bi-plane Simpson method was performed using a Vevo 2,100 system (Visual Sonics Inc.,Toronto,Canada) equipped with a linear scanner(30 MHz),as previously research[26].Briefly,mice were under 2%-isoflurane anesthesia,and brightness-mode movies of the parasternal long axis were acquired to mesure the length of ventricular.Mid-ventricular,apical,and basal short-axis views were acquired to calculate the area.The end-diastolic volume (EDV) and end-systolic volume (ESV) were calculated by the formula:V=(area mid-ventricular+area apical+area basal) × ventricular length/3.Ejection fraction (EF) was calculated as(EDV-ESV)/EDV×100%.To ensure the precision and reliability of experimental findings,two experienced cardiologists,blinded to genotype and treatment allocation,independently assessed myocardial function.
2.13.Measurement of infarct size
Infarct size was measured 24 h after I/R via Evans-blue (3%;Sigma-Aldrich) and 2,3,5-triphenyltetrazolium chloride (TTC;1%;Sigma-Aldrich) staining.For the Evans-blue staining,the thread around the LAD was re-ligated,and then the aorta was clamped using hemostatic forceps.The Evans-blue solution was retrogradeinjected from the aortic root.This solution stains non-ischemic areas of the heart blue,except for the anterior descending ramus.The heart was removed,stored at -20°C for 30 min,and then longitudinally sectioned.The sections were then incubated in 1%(V/V) TTC solution for 30 min at 37°C.Heart sections were photographed by a mirrorless camera (EOS R) equipped with 100 mm micro-lens.The lighting was performed by two flash lights to ensure a stable color temperature (5226.85°C).For the I/R heart,blue-and red-stained regions are non-ischemic and ischemic noninfarct myocardial (AAR) regions,respectively,and unstained regions correspond to ischemic infarct myocardium.AAR and infarction sizes were assessed using ImageJ.
2.14.Terminal deoxynucleotidyl transferase-mediated dUTP nickend labeling (TUNEL) assay
TUNEL assay was performed to detect the apoptotic cells in AAR 24 h after I/R.To this end,a one-step TUNEL assay kit(Beyotime Institute of Biotechnology,Shanghai,China) was employed on myocardial sections according to the manufacturer's instructions.Nuclei were then stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min.The samples were analyzed via a video camera connected to an Olympus IX-71 microscope,and the percentage of TUNEL+stain cells was calculated by using ImageJ software.
2.15.Masson's trichrome staining
Masson's trichrome staining was performed to analyze the sizes of the fibrotic scars 7 days post-I/R,following the manufacturer's protocol.Briefly,heart sections were subjected to deparaffinization and rehydration,and then incubated in Bouin's solution at room temperature overnight.Subsequently,they were stained at room temperature with hematoxylin for 30 min,Biebrich scarlet,phosphotungstic/phosphomolybdic acid,and aniline blue for 5 min,respectively,and 1% (V/V) acetic acid solution for 3 min.Finally,they were photographed and scar sizes were calculated using ImageJ software.
2.16.Enzyme-linked immunosorbent assay (ELISA)
The activity of oxoglutarate dehydrogenase complex (OGDHc,A101162,Fusheng Biotechnology,Shanghai,China),pyruvate dehydrogenase phosphatase (PDH,AS6321143,Fusheng Biotechnology),nicotinamide adenine dinucleotide phosphate (NADPH)oxidase (NADPH oxidase assay kit,H327-1;Jiancheng,Nanjing,China),cyclooxygenases (COX,COX activity assay kit,H210-1-2;Jiancheng),and xanthine oxidase (XOD,XOD assay kit,A002-1-1;Jiancheng) were quantified using specific ELISA kits.Samples from mice were extracted from the AAR 24 h after I/R.They were then homogenized in ice-cold PBS followed by centrifugation at 1,000gfor 10 min at 4°C.The supernatant was collected and used in sandwich ELISA following the protocols of the manufacturers.
Likewise,the BH4 level was determined using an ELISA kit(MY05696;Meiyi,Shanghai,China),and uncoupled NO synthase was quantitated by calculating the ratio of BH4 level/oxidized biopterins.cTnT and creatine kinase muscle and brain isoenzyme(CK-MB) levels were determined using ELISA (SEKM-0150 and SEKM-0152,respectively;Solarbio,Beijing,China).To this end,blood was collected from the abdominal aorta and centrifuged at 3,000gfor 10 min at 4°C.The supernatant was collected,and cTnT and CK-MB levels were measured according to the instructions provided by the kits.The absorbance of the samples at 450 nm was measured,and cTnT and CK-MB levels were calculated according to the regression equation.
2.17.Q-PCR
Total RNA was extracted by using the TRIzol method(Invitrogen,Carlsbad,CA,USA).RNA purity was assessed by using a nanodrop(Thermo Scientific Inc.,Waltham,MA,USA),and complementary DNA (cDNA) synthesis was then performed using a cDNA kit(Thermo Scientific Inc.).Q-PCR was run on a BioRad thermocycler machine.PCR primers for theTrpcsfamily,Ncx1,andCacna1cwere designed and optimized using PrimerDesign©(Table S1).Nontemplate control and reverse-transcriptase control were applied to verify the lack of DNA contamination,and primer specificity was analyzed by melt curves.Raw threshold cycle values were normalized to that of the housekeeping gene.
2.18.Quantitation of ROS
A dihydroethidium (DHE) kit (BestBio,Shanghai,China) was used for detecting ROS in heart sections.Heart sections from mice subjected to myocardial I/R were prepared 24 h after the reperfusion.The sections were stained with 10 μM DHE in a light-protected humidified chamber at 37°C for 15 min,and the overall ROS level was detected by measuring the total DHE fluorescence.
In addition,ROS generation in AMVMs and HL-1 cells was monitored using a MitoTracker Red(MTR)CM-H2XRos kit(Thermo Fisher Scientific Inc.).The cells subjected to H/R were incubated with 250 nM MTR for 30 min at 37°C.Afterward,fluorescence images were captured using an Olympus IX-71 microscope,and ImageJ was used for quantitative analysis.
2.19.Isolation of mitochondria
Mitochondria were isolated following established protocols[27]at 4°C.The hearts were extracted and then placed in a mitochondrial isolation buffer (mannitol-ethylene glycol tetraacetic acidsucrose-Hepes (MESH) buffer) composed of 220 mM mannitol,1 mM ethylene glycol tetraacetic acid,70 mM sucrose,and 10 mM Hepes at pH 7.4.The hearts were diced,rinsed to remove the blood,minced,and further homogenized in MESH buffer.The homogenates were centrifuged at 3,000gfor 10 min,and the pellet was resuspended in 10 mL of MESH buffer and subjected to centrifugation at 800gfor 10 min.The supernatant was then centrifuged at 3,000gfor 10 min.The resulting mitochondrial pellet was resuspended in 0.1 mL of MESH buffer,and the total protein concentration was calculated using the Bradford assay.
2.20.Measurement of O2·-/H2O2 release
Mitochondrial O2·-/H2O2release was assessed using the Amplex Ultra Red assay (AUR;Thermo Fisher Scientific Inc.) according to a previous report [28].Cardiac mitochondria were diluted to 0.1 mg/mL in MESH buffer and added to the wells of a black 96-well plate.AUR reagent was then added to each well at a final concentration of 10 μM.Reactions were started by adding 50 μM 2-oxoglutarate and 50 μM pyruvate,and 50 μM malate was added to complete the Krebs cycle,ensuring the full oxidation of 2-oxoglutarate to measure the ROS releases by OGDHc.Changes in fluorescence were monitored at the excitation and emission wavelengths of 565 nm and 600 nm,respectively,using a Spectramax M2 microplate reader(Molecular Devices,Shanghai,China)for 5 min.
2.21.Small interfering RNAs (siRNAs) transfection
Cells were transfected with specific siRNAs (synthesized by Hippo Biotechnology Co.,Hangzhou,China) targetingOgdhl,Trpc1,Trpc6,CaN,nuclear factor of activated T cells(Nfat),andNF-κB.A scrambled siRNA was applied as a negative control and produced similar results to those of untreated controls.Lipofectamine (Invitrogen) was mixed with 320 nM siRNA in OptiMEM (Gibco) and incubated for 20 min.The lipofectamine/siRNA complex was then added to the culture medium at a final concentration of 160 nM,and then the cells were incubated for 48 h.The siRNAs are listed in Table S2.
2.22.Plasmid construction and transfection
MouseTrpc1,Ogdhl,andNF-κBfull-length cDNAs were amplified using PCR with specific primers and then cloned into the pcDNA3.1 vector (Invitrogen;Table S3).HL-1 or HEK-293 were seeded in 6-wells or 24-wells culture dishes and allowed for adhered overnight.For transfection,pcDNA3.1(2.0 μg)and Lipofectamine 2,000(3 μL;Invitrogen) were separately diluted in serum-free OptiMEM(Gibco) to final volumes of 250 μL,mixed,incubated at 25°C for 15 min,and then added to the cell culture medium and incubated for 5 h at 37°C.Then the medium was replaced by DMEM containing 10% (V/V) FBS.
2.23.Luciferase assay
HEK293 cells were transfected with the mouseOgdhlpromoter andNF-κBexpression plasmid and cultured in 24-well plates for 24 h.Lysates were separated and luciferase activity was measured by the Dual-Luciferase Reporter Assay System (E1910;Promega Corporation,Cheyenne,WI,USA).The primer sequences are listed in Table S4.
2.24.Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using a SimpleCHIP®Enzymatic Chromatin IP Kit (Magnetic Beads) (9003;Cell Signaling Technology,Boston,MA,USA)according to the manufacturer's instructions.The NF-κB p65 (D14E12) XP®rabbit monoclonal antibody was obtained from Cell Signaling Technology.Fragments of the mouseOgdhlpromotor were amplified using PCR.The primer sequences are listed in Table S5.
2.25.Western blot assay
For in vivo experiments,protein samples were extracted from the AAR at the indicated time points post-I/R.For in vitro experiments,cells were collected at the indicated time points.Samples were homogenized in ice-cold radioimmunoprecipitation assay buffer containing protease inhibitors and then centrifuged at 4,000gfor 10 min at 4°C.The supernatants were then collected,and their total-protein concentrations were assessed by using a bicinchoninic acid assay kit(Leagene Biotechnology,Beijing,China).Equal amounts of samples were then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes.After blocking with defatted milk,the membranes were incubated with the following primary antibodies: mouse anti-β-actin (1:1,000;Sigma-Aldrich),rabbit anti-TRPC1 (1:1,000;Alomone Labs,Jerusalem,Israel),rabbit anti-TRPC6 (1:1,000;Abcam,London,UK),rabbit anti-p-NF-κB p65(1:1,000;Cell Signaling Technology),rabbit anti-NF-κB-p65(1:1,000;Cell Signaling Technology),rabbit anti-IκB-α(1:1,000;Cell Signaling Technology),rabbit anti-p-IκB-α (1:1,000;Cell Signaling Technology),rabbit anti-OGDHL (1:500;Affinity Biosciences,Trenton,NJ,USA),rabbit anti-CaNα (1:1,000;Cell Signaling Technology),and rabbit anti-CaNβ (1:500;Cell Signaling Technology).Then horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence were added to detect the target bands.Images were documented via densitometry (Bio-Rad,Carlsbad,CA,USA),and quantification was performed using ImageJ software.
2.26.Statistical analysis
Data are presented as mean ± standard error of means unless otherwise indicated.The choice of statistical calculation method was based on the number of groups to be compared.For comparison between any two groups,a Student'st-test was performed.Statistical significance for comparisons between more than two independent groups was tested using a one-way analysis of variance(ANOVA),followed by a Dunnettt-test.For analyses involving multiple influencing variables,a two-way ANOVA,followed by a Dunnettt-test,was performed.P-values <0.05 were considered to be statistical significance.
3.Results
3.1.Cardiac TRPC1 expression and activity are induced postmyocardial-I/R
Myocardial I/R (30 min/24 h) was induced in mice,and the messenger RNA(mRNA)level of eachTrpcwas measured via q-PCR.As shown in Fig.1A,Trpc1andTrpc6were the most abundantly expressed members of the TRPC family,and theTrpc5andTrpc7mRNAs were not at detectable levels.I/R resulted in the upregulation ofTrpc1andTrpc6in the AAR of the heart,compared with the levels in the sham group.The expression of otherTrpcswas not changed post-myocardial-I/R.Western blot results confirmed that in the mice subjected to myocardial ischemia for 30 min and reperfusion for various periods,cardiac TRPC1 and TRPC6 levels were progressively increased in correlation with the duration of reperfusion (Fig.1B).TRPC1 (Fig.S3A) and TRPC6 (Fig.S3B) levels were also increased in AMVMs subjected to H/R.

Fig.1.Canonical transient receptor potential channel(TRPC)1 expression and activity are increased post-myocardial ischemia/reperfusion(I/R).(A)Quantitative polymerase chain reaction to assess the expression of Trpcs,Cacna1c,and Na+/Ca2+exchanger 1(Ncx1)messenger RNA(mRNA)in area at risk(AAR)dissected from mice underwent myocardial I/R or sham operation.Expression levels were normalized to 18S RNA and expressed as percentage relative to 18S RNA (n=6 per group).(B) Representative Western blot images and relative quantification of TRPC1(left)and TRPC6(right)in the area at risk of mice undergoing I/R or sham operation(n=4 per group).(C)HL-1 cells were transfected with indicated small interfering RNAs(siRNA)for 48 h and underwent hypoxia/reoxygenation(H/R,6 h/9 h).TRPC channel-mediated Ca2+entry was assayed in the presence of TRPC agonist oleylacetyl-glycerol(OAG,10 μM)and cyclopiazonic acid(CPA,5 μM).Where indicated,SKF96365(SKF,5 μM)or the L-type Ca2+channel inhibitor nifedipine(Nif,10 μM)were used.(D)HL-1 cells were infected with the Trpc1 plasmid and assayed for TRPC-mediated Ca2+ entry.**P <0.01.BLQ: below the limit of quantification;Con: control.
Co-treatment of cardiac myocytes with CPA and OAG is widely used to analyze TRPC-mediated Ca2+entry into cells [29,30].To study whether TRPC1 and TRPC6 are involved in TRPC-mediated Ca2+influx in mice post-myocardial-I/R,we used this method to quantitate the Ca2+influx in HL-1 cells subjected to H/R.HL-1 cells displayed no detectable TRPC-mediated Ca2+entry under normoxic conditions(Fig.S4)but substantial TRPC-mediated Ca2+influx upon H/R (6 h/9 h).The specificity of TRPC-mediated Ca2+influx was validated using the TRPC antagonist SKF96365[31].The Ca2+influx was not inhibited by the LTCC antagonist nifedipine,documenting that the influx was independent of LTCC-mediated Ca2+influx.UnlikeTrpc6knockdown,knocking downTrpc1diminished the TRPC-mediated Ca2+influx in HL-1 cells subjected to H/R (Fig.1C).Additionally,HL-1 cells withTrpc1overexpression displayed increased TRPC-mediated Ca2+entry compared with the level in the control,and this effect was inhibited by SKF96365 (Fig.1D).These data suggest that TRPC1 is upregulated after myocardial I/R and plays a critical role in TRPC-mediated Ca2+influx.
3.2.Trpc1 deficiency ameliorates myocardial I/R injury
To clarify the roles of TRPC1 and TRPC6 in myocardial I/R,Trpc1-/-mice andTrpc6-/-mice were generated and then subjected to myocardial I/R injury.There was no difference in echocardiographic baseline among WT,Trpc1-/-,andTrpc6-/-mice before myocardialI/R (Table S6).Cardiac function post-I/R was assessed by calculating the EF via the bi-plane Simpson method (Fig.2A).The results showed that the absence ofTrpc1,unlike the absence ofTrpc6,significantly inhibited the EF decrease induced by the myocardial I/R injury,demonstrating thatTrpc1deficiency improved I/R-induced cardiac contractile dysfunction (Fig.2B).Additionally,the serum levels of cardiac injury biomarkers were measured.Trpc1-deficient mice displayed significantly decreased serum Troponin T (Fig.S5A)and CK-MB (Fig.S5B) levels post-I/R compared with those of WT mice.Results from TTC staining indicated thatTrpc1-/-mice had significantly smaller infarcts than the WT andTrpc6-/-mice after myocardial I/R (Figs.2C,2D,and S6).The hematoxylin and eosin(H&E) staining results indicated that WT andTrpc6-/-mice displayed disruption of myocardial fibers,tissue edema,and neutrophil infiltration in the AAR of the left ventricle myocardium after myocardial I/R.All these injuries were relatively less intense inTrpc1-/-mice subjected to myocardial I/R(Figs.2E and S7).We also assessed for cardiac fibrosis 7 days post-myocardial-I/R by using Masson's trichrome staining and found thatTrpc1-/-mice displayed smaller fibrotic scars than WT andTrpc6-/-mice (Fig.S8).Since apoptosis is a hallmark cellular event post-myocardial-I/R,TUNEL staining was performed to evaluate the level of associated apoptosis.Fewer cells (19.7% ± 4.2%) in the AAR underwent apoptosis in theTrpc1-/-mice subjected to myocardial I/R than in the WT(42.0%± 6.2%) orTrpc6-/-(37.6% ± 5.1%) counterparts (Figs.2F,2G,and S9A).The level of lactate dehydrogenase(LDH)release was then determined to evaluate the level of associated necrosis[32].The total LDH release in the hearts ofTrpc1-/-mice submitted to I/R was significantly less than that in the WT counterparts (Fig.S9B).Collectively,these results suggest that TRPC1 plays a critical role in myocardial I/R injury in mice.

Fig.2.Knockout of canonical transient receptor potential channel 1 (Trpc1) attenuates myocardial ischemia/reperfusion (I/R) injury.(A) Representative echocardiographs recorded from mice post-I/R(30 min/24 h).End-diastolic volume(EDV)and end-systolic volume(ESV)were determined by the bi-plane Simpson method requires the length of the ventricle(red)obtained from a parasternal long-axis(PSLAX)view(endocardial tracing is blue),and the left ventricular areas from three orthogonal short-axis(SAX)views(midventricular,apical,and basal;blue disks).(B) Quantitative analysis of cardiac ejection fraction (EF) in wild type (WT), Trpc1-/-,and Trpc6-/- mice underwent sham operation or I/R (30 min/24 h)calculated as(EDV-ESV)/EDV×100%(n=5 per group).(C)2,3,5-triphenyltetrazolium chloride-Evans blue staining from WT,Trpc1-/-,and Trpc6-/-mice underwent sham operation or I/R(30 min/24 h).For I/R mice,white indicates infarct area,red indicates area at risk(AAR),and blue indicates the tissue not subjected to I/R injury.(D)Average data showing the percentages of the infarct area to AAR (n=5 pairs of mice).(E) Representative hematoxylin and eosin (H&E) staining images of the AAR from WT, Trpc1-/-,and Trpc6-/-mice after I/R(30 min/24 h).(F)Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling(TUNEL,green)and α-actin(red)staining was used to observe the apoptosis myocytes in the AAR post-I/R (30 min/24 h).4′,6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei (blue).(G) Quantification of apoptosis cells of heart sections (n=4 per group).**P <0.01.Con: control;IS: infarct size.
3.3.TRPC1 expression in cardiomyocytes contributes to myocardial I/R injury
Considering the role of TRPC1 in I/R-induced cardiomyocytes injury and Ca2+disorder,we then employed an AAV9 vector with a cTnT promoter to selectively knockdownTrpc1in cardiomyocytes to explore the specific contribution of cardiomyocyte-specific expression ofTrpc1to myocardial I/R progression (Fig.3A).AAV9-cTnT-shTrpc1 or AAV9-cTnT-shControl (shCon) injection did not cause any change in the echocardiography baseline in the absence of myocardial I/R(Table S7).The results from Western blot showed that the AAV9-cTnT-shTrpc1 delivery successfully knocked down the TRPC1 expression post-myocardial-I/R(Fig.3B).The outcome of myocardial I/R was then evaluated based on cardiac functions(Fig.3C),infarct size (Figs.3D and E),and apoptotic cells (Figs.3F,3G,and S10).All these results support that the cardiomyocytespecific silencing ofTrpc1replicated the protective effect of globally knocking out TRPC1,suggesting that cardiomyocyte-specific expression of TRPC1 is involved in myocardial I/R injury.
3.4.Trpc1 deficiency attenuates cardiomyocyte injuries induced by simulated I/R
To further prove the role of TRPC1 in simulated I/R in vitro.We first investigated whether TRPC1 was involved in the Ca2+influx to cardiomyocytes post-myocardial-I/R.AMVMs were isolated from the hearts of WT,Trpc1-/-,andTrpc6-/-adult mice that underwent sham operation or myocardial I/R,and the Ca2+influx was quantitated.The AMVMs from the sham-operated mice showed no detectable TRPC-mediated Ca2+influx (Fig.S11),whereas those from the WT mice subjected to post-myocardial-I/R showed a substantial Ca2+influx (Figs.4A and S12).Notably,this Ca2+influx was diminished in AMVMs isolated fromTrpc1-/-mice,unlike in those isolated fromTrpc6-/-mice postmyocardial-I/R (Fig.4A).
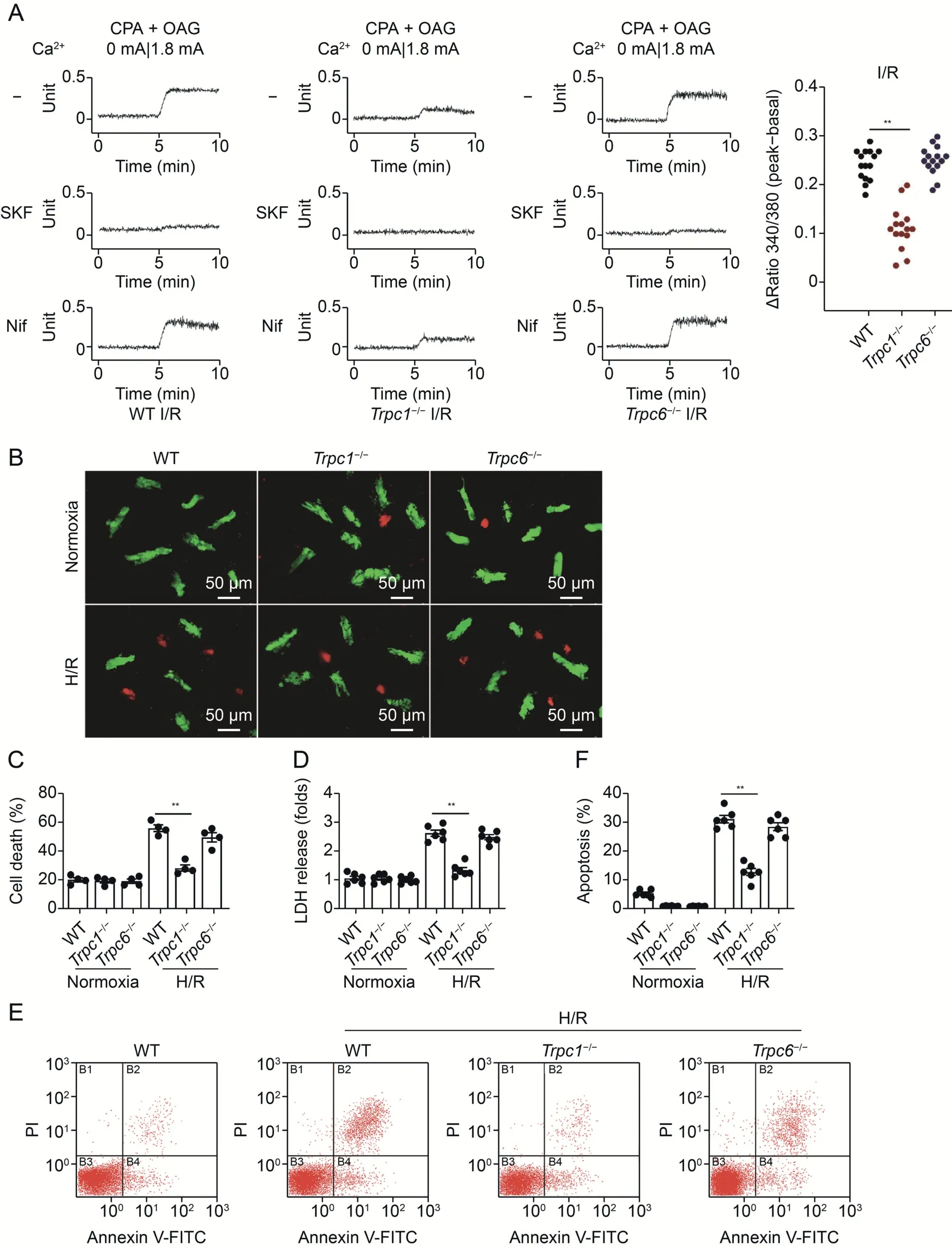
Fig.4. Canonical transient receptor potential channel 1 (Trpc1)deficiency attenuates simulated ischemia/reperfusion(I/R)induced injury in primary cultures of cardiomyocytes.(A)Adult mice ventricular myocytes(AMVMs)were isolated from wild-type(WT),Trpc1-/-,and Trpc6-/-mice post-myocardial I/R.TRPC channel-mediated Ca2+entry was assayed in the presence of oleyl-acetyl-glycerol(OAG,10 μM)and cyclopiazonic acid(CPA,5 μM).SKF96365(SKF,5 μM)or the L-type Ca2+channel inhibitor nifedipine(Nif,10 μM)were used.(B) AMVMs isolated from WT, Trpc1-/-,and Trpc6-/- mice were exposed hypoxia/reoxygenation (H/R,5 h/1 h) condition.Live/dead assays were used to assess cell viability.(C)Quantitative data of AMVMs death.Total numbers of live cytosol(green)and dead nuclei (red)were counted in low-power fields (n=4 per group).(D)Relative secreted levels of lactate dehydrogenase (LDH) in the different groups of AMVMs (n=6 per group).(E) Flow cytometry images of Annexin V-propidium iodide (PI) staining in neonatal mice ventricular myocytes underwent H/R injury.(F) Quantitative analysis of flow cytometry results (n=3 per group).**P <0.01.FITC: fluorescein isothiocyanate.
We then studied whether TRPC1 was involved in H/R-induced cardiomyocyte injury.AMVMs were isolated from the hearts of adult WT,Trpc1-/-,andTrpc6-/-mice,and then the cells were subjected to H/R (5 h/1 h).Live/dead viability assay,in which the live cells were stained with calcein AM(green)and the nuclei of the dead cells were stained with ethidium homodimer-1 (red),demonstrated that H/R significantly induced the death of AMVMs,compared with the normoxic level,and upon H/R,Trpc1-/-but notTrpc6-/-AMVMs displayed enhanced cell viability compared with that of WT AMVMs (Figs.4B and C).Trpc1-/-but notTrpc6-/-AMVMs also displayed decreased LDH release compared with that of WT AMVMs (Fig.4D).The MTT and LDH assays were then performed on NMVMs subjected to H/R.In WT andTrpc6-/-NMVMs,H/R(6 h/9 h)decreased the cell viability and enhanced the release of LDH,butTrpc1-/-NMVMs displayed enhanced cell viability(Fig.S13A)and reduced LDH release(Fig.S13B)compared with the levels in the WT group.Furthermore,the apoptosis of cells was assessed via flow cytometry.WT NMVMs that underwent H/R exhibited high rates of apoptosis and necrosis;deficiency ofTrpc1,but notTrpc6,significantly inhibited the apoptosis induced by H/R injury (Figs.4E and F).In HL-1 cells,unlike the effect of knocking downTrpc6,Trpc1knock-down significantly suppressed the H/Rinduced apoptosis,compared with the rate in the control group(Fig.S14A).Furthermore,overexpression ofTrpc1in HL-1 cells increased the H/R-induced apoptosis (Fig.S14B).To further assess the impact of TRPC1 on HL-1 cells post-H/R,we assessed the mitochondrial permeability transition (mPT),a hallmark of H/R injury,by using a JC-1 reporter dye [33].Unlike theTrpc6knockdown,Trpc1knock-down in HL-1 cells dramatically attenuated the changes in mTP (Fig.S14C).Furthermore,overexpression ofTrpc1deteriorated the mPT trend upon H/R injury(Fig.S14D).These results suggest that TRPC1 contributes to cardiomyocyte injury post-simulated-I/R.
3.5.Trpc1 knockout is associated with a distinctive myocardial gene expression profile
To further investigate the role of TRPC1 in myocardial I/R injury,RNA-seq analysis was performed to identify the DEGs in the AAR of WT,Trpc1-/-,andTrpc6-/-mice 24 h after the I/R.Knocking outTrpc1affected the expression levels of 1,008 genes (731 upregulated and 277 downregulated),compared with the levels in WT mice,after I/R(Figs.5A and B).GO analysis was then performed to gain an overview of the nature of the DEGs and the shared pathways.Here,we listed the top 10 enriched clusters in the comparison between the WT-I/R andTrpc1-/--I/R transcriptomes.Interestingly,two terms,namely “ROS generation process” and“Response to oxidative stress,” were involved in the ROS levels post-myocardial-I/R (Fig.5C).However,these GO terms were not identified upon the comparison of WT andTrpc6-/-mice subjected to I/R(Fig.S15).Thus,we speculated that TRPC1 plays an important role in regulating ROS generation post-myocardial-I/R.
3.6.Trpc1 deficiency attenuates the post-I/R ROS upregulation
To determine the potential impact of TRPC1 on the postmyocardial-I/R ROS upregulation,we measured the ROS levels in the AAR of WT,Trpc1-/-,andTrpc6-/-mice after I/R by using a DHE kit.The heart sections from all the mice subjected to myocardial I/R had widespread and markedly intensified DHE fluorescence compared with that in the sham-operated groups.However,of the I/R-injured mice,theTrpc1-/-mice displayed a significant decrease in DHE fluorescence intensity unlike theTrpc6-/-mice,compared with the intensity in the WT mice (Figs.5D and S16).Aconitase enzyme activity is extremely sensitive to ROS and thus can serve as an indicator of ROS level [34].We observed that myocardial I/R injury caused a significant decrease in aconitase activity in the AAR,compared with the level in the sham group,indicating upregulation of ROS upon the I/R.However,knocking outTrpc1,unlike theTrpc6knock-out,suppressed the ROS upregulation (Fig.5E).In parallel,we assessed the ROS level in AMVMs subjected to H/R,by monitoring the reduced form of MTR.The AMVMs subjected to H/R displayed ROS upregulation;however,this effect was suppressed inTrpc1-/-AMVMs (Fig.5F).In HL-1 cells subjected to H/R,Trpc1knockdown caused a significant decrease in MTR fluorescence intensity (Fig.5G) and an increase in aconitase activity (Fig.5H),compared with the level in the control cells.These results suggest that TRPC1 upregulates ROS upon myocardial I/R.
3.7.Trpc1 deficiency attenuates the post-I/R ROS upregulation via downregulating α-ketoglutarate dehydrogenase
To further explore the mechanism by which TRPC1 upregulates ROS upon myocardial I/R,we screened 16 ROS-related genes that were substantially down-or up-regulated inTrpc1-/-mice subjected to I/R,compared with the levels in WT mice subjected to I/R.Among these genes,Ogdhl,the rate-limiting component of the mitochondrial multi-enzyme oxoglutarate dehydrogenase complex(OGDHc),was the most markedly downregulated gene (Fig.6A).These observations were further validated via real time (RT)-qPCR(Figs.6B and S17).Western blot experiments also showed that the OGDHL protein level was increased in the AAR upon I/R(Figs.6C and S18),and this increase was diminished inTrpc1-/-mice (Fig.6C).Depletion ofOgdhlwas reported to reduce OGDHc activity,which is vital for Krebs cycle metabolism and accounts for a substantial fraction of the ROS release in the cardiac tissue[35].Therefore,the OGDHc enzyme activity was measured,andTrpc1-/-mice were found to display lower enzyme activity than WT mice post-I/R.However,the level of PDH,another vital enzyme for the Krebs cycle and source of ROS[36],was unaffected inTrpc1-/-mice(Fig.6D).NADPH oxidase,COX,XOD,and uncoupled NO synthase also contribute to ROS generation post-myocardial-I/R[37].The activity of these enzymes in the AAR was assessed,and the results showed thatTrpc1deficiency slightly decreased the NADPH oxidase activity post-I/R (Fig.S19A) but did not affect the activity of COX,XOD,or uncoupled NO synthase (Figs.S19B-D).Thus,Trpc1knockout mainly affects the OGDHc among the enzymes related to ROS generation.

Fig.6.Canonical transient receptor potential channel 1 (TRPC1) increases reactive oxygen species (ROS) generation via regulating oxoglutarate dehydrogenase-like (Ogdhl)expression.(A)Heatmap of the ROS related gene profiles in the area at risk(AAR)of wild-type(WT)and Trpc1-/-mice that underwent myocardial I/R(30 min/24 h).(B-D)The AAR region was separated 24 h after I/R in WT or Trpc1-/- mice.The (B) messenger RNA (mRNA),and (C) protein levels of OGDHL were determined by quantitative polymerase chain reaction (q-PCR) or Western blot,respectively.(D) Oxoglutarate dehydrogenase complex (OGDHc) and pyruvate dehydrogenase phosphatase (PDH) activities were detected by enzyme-linked immunosorbent assay (ELISA) kit (n=3 per group).(E) ROS generation was determined in the HL-1 cells transfected with a green fluorescent protein targeting control(Con)or Ogdhl,lectin galactoside binding soluble 3(Lgals3),tripartite-motif protein 30(Trim30d)small interfering RNAs(siRNAs),then underwent hypoxia/reoxygenation(H/R,6 h/9 h)(n=3 per group).(F)The mRNA and(G)protein levels of OGDHL and(H)OGDHc activity in WT or Trpc1-/-adult mice ventricular myocytes upon H/R(5 h/1 h)condition(n=4 per group).(I)O2·-/H2O2 release was examined in mitochondria prepared from the cardiac tissue oxidizing 2-oxoglutarate.Mitochondria(0.1 mg/mL)was diluted in reaction buffer containing 2-oxoglutarate(50 μM)and the production of H2O2 was measured via Amplex Ultra Red(n=5).(J)Mitochondria prepared from the cardiac tissue 24 h post-I/R was treated with OGDHc inhibitor,3-methyl-2-oxovaleric acid(KMV,10 mM),or Complex III inhibitor,myxothiazol(MYX,10 mM),for 30 min,and then diluted in reaction buffer containing 2-oxoglutarate(50 μM).Production of H2O2 was measured via Amplex Ultra Red(n=6).(K)Representative images and quantitative analysis of MitoTracker Red(MTR)fluorescence intensity in HL-1 cells transfected with Trpc1 siRNA and Ogdhl plasmid and underwent H/R (6 h/9 h) then loaded with MitoTracker dye (250 nM) (n=3 per group).**P <0.01.n.s.: no significance.
To further explore the role of OGDHL in the ROS generation postmyocardial-I/R,we knocked down the ROS-related genes in HL-1 cells and found that onlyOgdhlknock-down remarkably suppressed the H/R-induced ROS upregulation (Fig.6E).We also observed thatTrpc1-/-AMVMs displayed significant decreases inOgdhlmRNA(Fig.6F)and protein(Figs.6G and S20)levels as well as OGDHc activity (Fig.6H),compared with those in WT AMVMs,upon H/R.Trpc1knock-down and overexpression in HL-1 cells suppressed (Figs.S21A and B) and enhanced (Figs.S21C and D),respectively,the mRNA and protein levels ofOgdhl,compared with those in WT HL-1 cells,upon H/R.OGDHc has been reported to generate ROS via oxidizing 2-oxoglutarate[28].Hence,we isolated the mitochondria of the cardiac tissue from mice,and the ROS release rates from OGDHc were measured.As shown in Fig.6I,OGDHc-generated ROS level was significantly lower in the cardiac mitochondria isolated fromTrpc1-/-mice than in those from WT mice post-myocardial-I/R.Pre-treatment of mitochondria with the OGDHc inhibitor,3-methyl-2-oxovaleric acid(KMV,10 mM),unlike the pre-treatment with a Complex III inhibitor,myxothiazol(MYX,10 mM),abolished this effect (Fig.6J).To investigate the role of OGDHc in TRPC1-mediated ROS generation,we overexpressedOgdhlinsiTrpc1-treated HL-1 cells and found that theTrpc1knockdown decreased the H/R-induced ROS upregulation in HL-1 cells,and overexpression ofOgdhlreversed this decrease(Fig.6K).These results indicate that TRPC1 upregulates ROS via OGDHL postmyocardial-I/R,suggesting thatOgdhlplays a critical role in the TRPC1-mediated ROS generation after myocardial I/R.
To further investigate whetherTrpc1deficiency ameliorated myocardial I/R injury via OGDHL,Ogdhlwas overexpressed in cardiomyocytes ofTrpc1-/-mice via intra-myocardial injection of AAV9 particles expressingOgdhl.The myocardial I/R injury outcome was evaluated by assessing cardiac function and performing TTC/Evansblue staining.The results showed that AAV-Con-treatedTrpc1-/-mice displayed higher EF than AAV-Con-treated WT mice,but this trend was diminished in AAV-Ogdhl-treatedTrpc1-/-mice(Fig.S22A).The results from TTC staining showed that the AAV-ContreatedTrpc1-/-mice had significantly smaller infarcts than the AAV-Con-treated WT mice post-I/R.Conversely,this trend was reversed in AAV-Ogdhl-treatedTrpc1-/-mice (Figs.S22B and C).These results indicate that the cardioprotective effect ofTrpc1knockout is counteracted byOgdhloverexpression.
3.8.TRPC1 regulates Ogdhl expression through the Ca2+/CaN/NF-κB signaling
Previous findings have reported that NF-κB regulated genes may play critical roles in regulating ROS levels in various cell types[38].Thus,we speculated that TRPC1 regulatedOgdhlafter I/R via the NF-κB signaling pathway.To test this hypothesis,the levels of p-NFκB p65 and p-IκB-α in AMVMs were measured using Western blot.Trpc1-/-AMVMs displayed significant downregulation of p-NF-κB p65 (Figs.7A and S23) and p-IκB-α (Figs.7B and S24),compared with the levels in WT AMVMs,upon H/R.Thus,TRPC1 positively regulated NF-κB after myocardial I/R.Furthermore,knocking down NF-κB in HL-1 cells dramatically decreased the H/R-induced increase in the OGDHL mRNA and protein levels,as assessed via RTqPCR and Western blot,respectively (Figs.7C,7D,and S25).Although NFAT is a key transcriptional activator involved in heart diseases,knocking it down did not affect the H/R-inducedOgdhlupregulation (Figs.7C and D).Silencing NF-κB also dramatically decreased the ROS generation induced by H/R injury (Fig.S26).Furthermore,the protective effect ofTrpc1deficiency against I/R was counteracted by overexpressing NF-κB in cardiomyocytes,as demonstrated by cardiac function evaluation and TTC/Evans-blue staining (Fig.S27).Taken together,these results indicate that TRPC1 upregulatesOgdhland aggravates I/R injury through the NFκB signaling pathway.To further investigate how NF-κB upregulatesOgdhl,we performed a series of experiments on HEK293 cells.First,we predicted the possible binding site of NF-κB on theOgdhlpromotor sequence and their relative positions in silico by using the JASPAR database.The results showed that NF-κB might interact with the GGGAGAGCCC sequence at the 438-447 promoter terminal region ofOgdhl(Table S8).Then,HEK293 cells were transfected with an NF-κB expression or control plasmid,and theOgdhlmRNA level was measured via RT-qPCR.We observed that theOgdhlmRNA level was increased by the transfection of the cells with the NF-κB expression plasmid (Fig.7E).Additionally,the luciferase assay revealed that NF-κB overexpression increased the activity of theOgdhlpromoter (Fig.7F).Next,we conducted ChIP assays to detect whether NF-κB could directly bind to the promotor ofOgdhland observed that NF-κB p65 overexpression caused an increase in its binding to the promotor ofOgdhl(Fig.7G).These observations indicate that NF-κB directly binds to theOgdhlpromotor,resulting in transcriptional activation ofOgdhl.
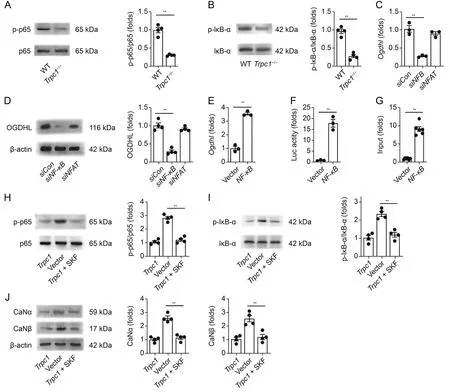
Fig.7.Canonical transient receptor potential channel 1 (TRPC1) regulates oxoglutarate dehydrogenase-like (Ogdhl) via Ca2+/calcineurin (CaN)/nuclear factor kappa-B (NF-κB)signaling pathway.(A,B)Representative Western blot images and relative quantification of p-NF-κB p65/NFκB p65(p65)(A)and p-IκB-α/IκB-α(B)in wild-type(WT)and Trpc1-/-adult mouse ventricular myocytes upon hypoxia/reoxygenation(H/R) (5 h/1 h)condition (n=4 per group).(C) The messenger RNA (mRNA) of Ogdhl in the HL-1 cells transfected with control(Con)or NF-κB small interfering RNA(siRNA)upon H/R(6 h/9 h)(n=3 per group).(D)Representative Western blot images and relative quantification of OGDHL in the HL-1 cells transfected with control or NF-κB siRNA,and then underwent H/R (6 h/9 h) (n=4 per group).(E) The mRNA of Ogdhl after transfected with the NF-κB expression or control plasmid (n=3 per group).(F) HEK-293 cells were transfected with NF-κB expression plasmid and Ogdhl promotor luciferase reporter constructs.Luciferase activity was detected 36 h after transfection using a betascope analyzer (n=3 per group).(G) The association of NF-κB with the Ogdhl promoter was analyzed by a quantitative chromatin immunoprecipitation(ChIP)assay(n=6 per group).Representative Western blot images and relative quantification of(H)p-NF-κB p65/NF-κB p65 and(I)p-IκB-α/IκB-α in the HL-1 cells transfected with vector or Trpc1 and then treated with vehicle or SKF96365(n=4 per group).(J)Representative Western blot images and relative quantification of CaNα and CaNβ in the HL-1 cells transfected with vector or Trpc1 and then treated with a vehicle or SKF96365 (SKF) (n=5 per group).**P <0.01.NFAT: nuclear factor of activated T cells.
We next investigated whether TRPC1 could regulate the NF-κB signaling via its Ca2+channel activity.HL-1 cells were transfected with aTrpc1expression plasmid and then treated with SKF96365 to block the TRPC-mediated Ca2+influx.The results indicated that SKF96365 influenced the p-NF-κB p65(Fig.7H)and p-IκB-α(Fig.7I)levels in the HL-1 cells transfected with the expressionTrpc1plasmid.These results suggest that TRPC1 regulates NF-κB likely dependent on its channel activity.CaN is a calcium-responsive phosphatase that has been reported to promote NF-κB nuclear translocation and transcriptional activity [39].Thus,we investigated whether TRPC1 regulated NF-κB through the Ca2+/CaN pathway,and the results indicated that overexpression ofTrpc1increased the CaN protein level in HL-1 cells.This increase,however,was diminished by SKF96365(Fig.7J).Furthermore,inhibition of CaN decreased the ROS generation in HL-1 cells upon H/R injury(Fig.S28).Taken together,these results suggest that TRPC1 regulates OGDHL-and OGDHc-mediated ROS generation via the Ca2+/CaN/NF-κB signaling pathway.
4.Discussion
Myocardial I/R injury is one of the leading causes of cardiac injuries and deaths.Therefore,it is of urgent demand to explore the underlying molecular mechanisms.In this study,we demonstrated that TRPC1 was prominently upregulated and played a critical role in the Ca2+influx to cardiomyocytes post-myocardial-I/R.TRPC1 upregulated ROS and aggravated myocardial I/R injury via upregulating OGDHL.Importantly,these findings shed light on previously undiscovered functions of TRPC1 during myocardial I/R and provide new insights into the mechanism of myocardial I/R.
TRPC isoforms constitute a group of non-selective cation channels and play important roles in the pathological processes of diverse diseases[14].Alterations in cellular Ca2+homeostasis have been considered to contribute to the cell death caused by myocardial I/R[40].A novel observation of the present study is that TRPC1-mediated Ca2+was found to play a critical role in the pathological process of myocardial I/R injury.Alterations in the Ca2+homeostasis can also occur in both ischemia and reperfusion periods.Upon reperfusion,the myocardium can suffer additional damage,surpassing the injury caused by ischemia alone,leading to adverse outcomes such as myocyte death,and account for nearly 50%of the final infarct size[41].In our study,we found that TRPC1 was upregulated during the reperfusion pathological process.Moreover,the TRPC-mediated Ca2+influx was detectable postreperfusion,and hence TRPC may contribute to the myocardial injury caused by the reperfusion process.
To further investigate the potential involvement of TRPC1 in myocardial I/R injury,RNA-seq analysis was performed.The results showed that knocking out TRPC1 most significantly impacted GO categories associated with ROS.This cue is supported by the observation thatTrpc1deficiency reduced the I/R-induced ROS upregulation in vivo and in vitro.ROS are particular drivers of mPT and oxidative damage of intramitochondrial structures and molecules.ROS also contributes to post-infarction remodeling even beyond the acute setting [42].Thus,we hypothesized that TRPC1 deficiency attenuates myocardial I/R injury by suppressing ROS upregulation.The mechanism of ROS generation is complicated.Complexes I and III were used to be considered as the major sites of ROS generation in mitochondria [35].It is now recognized that mitochondria ROS can also be generated in other sites.Notably,PDH and OGDHc,which are enzymes of the Krebs cycle,have been reported to be the major contributors to the overall ROS yield in skeletal-muscle mitochondria [43].In our study,theOgdhlgene was the most markedly downregulated gene inTrpc1-/-mice,compared with the level in WT mice post-myocardial-I/R.Trpc1knockout also decreased OGDHL protein level and OGDHc activity but did not impact the PDH activity.Furthermore,knocking downOgdhlreduced the simulated-I/R-induced ROS upregulation,whereas overexpression ofOgdhlincreased the ROS generation inTrpc1knockdown cardiomyocytes subjected to simulate I/R.These findings are consistent with a recent study showing that OGDHc is a more significant source of ROS than PDH in the cardiac and hepatic tissues [44].Additionally,observations in neuronal mitochondria have indicated that OGDHc produces more ROS than PDH[45].Our results further showed that TRPC1 upregulated ROS via OGDHc post-myocardial-I/R.
We then investigated the signaling pathway underlying the upregulation ofOgdhlby TRPC1.A transduction pathway is required for the TRPC-controlled Ca2+microdomain transferring signal to the nucleus.For the Ca2+-dependent transcription-coupling signaling pathway,we specifically focused on the CaN signaling.The pathological role of the TRPC signaling in diseases such as cardiac hypertrophy has been repeatedly attributed to disturbances in the CaN signaling [29].In our study,overexpression of TRPC1 in cardiomyocytes upregulated CaNα and CaNβ,indicating that TRPC1-mediated Ca2+influx is sufficient to activate CaN.This finding aligns with an expanding body of evidence supporting that TRPC-mediated Ca2+influx serves as an important Ca2+source for the activation of CaN [9].These results may partly explain why TRPC-mediated Ca2+influx conferred the subsequent effects.The mechanistic coupling between TRPC1-CaN might take place in specific subcellular locations and cause dramatic effects on myocardial I/R injury.
It is well established that TRPC-CaN participates in cardiac pathological processes by activating transcriptional factors [30].Previous studies have demonstrated that NFAT and NF-κB are the most frequently involved transcription factors downstream of TRPCs [46].TRPC mediates cardiac hypertrophic response through NFAT,which is directly dephosphorylated by CaN and consequently translocates to the nucleus [47].NF-κB plays a major role in inflammation and immunity [48].NF-κB has previously been shown to regulate the expression of multiple genes involved in regulating ROS generation [38].To verify whether NFAT or NF-κB participates in the process of TPRC1-CaN regulatingOgdhl,we knocked down each of these genes and observed that 1) silencing NF-κB,unlike the NFAT knock-down,decreased theOgdhexpression in myocytes,whereas overexpression of NF-κB increasedOgdhexpression;2)Trpc1-/-AMVMs and siTrpc1-treated HL-1 cells displayed decreased p-NF-κB 65 levels compared with the WT level,upon simulated I/R;3) knocking down CaN or the pharmacological inhibitor of CaN (FK506) decreased p-NF-κB p65;and 4)ChIP and luciferase assays showed that NF-κB binds to theOgdhlpromotor,resulting in transcriptional activation ofOgdhl.These findings are supported by previous studies that showed that TRPC regulates hypertrophic scar contracture via NF-κB activation [49].Thus,our study suggests a causal link between TRPC1,CaN,NF-κB,andOgdhl,indicating that TRPC1 upregulatesOgdhlvia the NF-κB signaling rather than the NFAT pathway.
Here,a general concern is why a very small amount of Ca2+influx mediated by TRPC1 can have such dramatic and specific effects on cell signaling.Disruption of Ca2+homeostasis participates in the pathogenesis of myocardial I/R [40].Contractile Ca2+proteins,such as LTCC and NCX,mediate large amounts of Ca2+alteration within a cardiomyocyte [50].However,LTCC and NCX blockers have not shown any beneficial effect on survival in clinical trials [51-54].In addition to these contractile Ca2+proteins,cardiomyocytes are also equipped with some Ca2+-handling proteins controlling signaling pathways [5].Previous studies have reported that TRPCs upregulation or activation is critical for cardiac Ca2+signaling underlying cardiac hypertrophy [12].TRPC channels can generate a complex spatiotemporal Ca2+pattern [46].The architecture of the TRPC-Ca2+signaling domain has the potential to generate and regulate distinct intracellular Ca2+microdomains[13].This segregated Ca2+signal can be decoded for the regulation of specific downstream functions[55].For instance,TRPC-mediated Ca2+influx is sufficient and essential to activate signaling pathways,such as the CaN that plays a critical role in various cardiac diseases [56-59].Thus,it is plausible that a small amount of TRPC1-mediated Ca2+influx causes cardiomyocyte death post-I/R.
Previous studies have identified TRPC isoforms responsible for the Ca2+influx and its relationship with myocardial infarct[60-63].Interestingly,He et al.[60] have found thatTrpc3/6/7 knockout mice subjected to myocardial I/R exhibit smaller infarct size,improved cardiac function,and reduced cardiac damage than their WT counterparts.However,there are some significant differences between our study and theirs.The initial finding that led He et al.[60]to assess for the roles of TRPC3/6/7 in I/R injury was that TRPC3/6/7 deficiency diminished 60% of the store-operated Ca2+entry(SOCE)evoked by thapsigargin in NMVMs.Confusingly,previous studies have demonstrated that TRPC3/6/7 mainly participates in receptor-operated Ca2+entry rather than SOCE[64,65].TRPC1 has been consistently associated with SOCE in various cell types [66,67],but the impact of TRPC1 deficiency on SOCE has not been studied.Furthermore,in these studies,Ca2+-influx analyses were performed in NMVMs and H9C2 cardiomyoblasts under the normoxic condition,which may not reflect the role of TRPC channel in Ca2+alteration in myocytes upon I/R.Accumulated evidence shows that TRPC expression and activity change at different levels upon stimulation [14].Unlike the previous studies,the expression of each TRPC isoform was assessed post-myocardial-I/R in our study.Based on this finding,we focused on TRPC1 since it was the most upregulated isoform post-I/R.Then theTrpc1-/-mice were generated to study the exact contribution of TRPC1 to myocardial I/R injury,whereasTrpc3/6/7 triple knockout mice,which may not reflect the precise role of certain isoforms,were used in the previous studies.Meanwhile,to actually reflect the TRPC-mediated Ca2+alteration in cardiomyocytes subjected to I/R,we assessed the Ca2+influx by using the AMVMs isolated from the heart subjected to I/R.Differences in experimental designs might explain the differences between our results and theirs.
Previous studies have shown that TRPCs affect basic and cell type-specific functions of the major cell types of the cardiovascular system in addition to cardiomyocytes,such as endothelial cells,fibroblasts,and macrophages[68].In the present study,we focused on cardiomyocytes since they are the functional units of the heart and constitute the majority of the cardiac cell mass[69].It is worth noting that existing research indicates that the contribution of TRPC1 to the ischemic heart is not restricted to cardiomyocytes.Wen et al.[70] have reported that TRPC1 upregulation in endothelial cells increase angiogenesis and facilitates recovery post myocardial infarction.In their study,TRPC1 conditional knockout in endothelial cell decreased EF value,enlarged infarct size,and reduced capillary dencity in the infarct area post myocardial ischemia,suggesting that TRPC1 in endothelial play a critical role in stimulating angiogenesis after myocardial ischemia.Thus,the impact ofTrpc1deficiency on endothelial cells post-I/R needs to be further investigated.Meanwhile,we found that mice withTrpc1deficiency displayed smaller fibrotic scars than WT mice postmyocardial-I/R.Previous studies have demonstrated that TRPCs contribute to fibrosis post-myocardial-infarct[62,71].For instance,TRPC6 in fibroblasts participate in myofibroblast transformation,andTrpc6-/-mice show fewer myofibroblasts in the infarcted area[62].These results suggest that TRPC1 in fibroblasts may also influence the outcome of I/R.Additionally,in addition to serving as a nonselective cation channel,several studies have reported that TRPC1 confers its functions independently of its channel activity but by embedding in a protein complex with cell surface receptors or cytosolic proteins[67,72].For instance,TRPC1 acts as a scaffold at the cell surface to assemble a signaling complex to stimulate neurite outgrowth independently of a Ca2+influx[73].Thus,the impact of TRPC1 on myocardial I/R may involve other mechanisms.Our study suggests that inhibiting TRPC1 can mitigate cardiomyocyte injury induced by I/R.However,there is no specific inhibitor of TRPC1,because the amino acid sequences of different TRPC proteins are highly homologous[74],hampering the development of specific inhibitors of the TRPC1 isoform.Thus,in this research,Trpc1-/-mice were applied to study the precise role of TRPC1 in myocardial I/R injury.
5.Conclusions
In summary,the results of this study highlight the key molecular role of TRPC1 in the ROS upregulation following myocardial-I/R injury.TRPC1 exacerbates the oxidative injury caused by I/R through the upregulation of OGDHL via the Ca2+/CaN pathway.Targeting TRPC1 may provide a potential therapeutic strategy for mitigating the adverse effects of myocardial I/R.
CRediT author statement
Hui-Nan Zhang:Methodology,Validation,Formal analysis,Investigation,Writing -Original draft preparation,Reviewing and Editing,Visualization;Meng Zhang:Conceptualization,Resources,Methodology,Investigation,Validation;Wen Tian:Writing -Reviewing and Editing,Methodology,Validation,Formal analysis;Wei Quan:Methodology,Validation,Investigation,Writing -Reviewing and Editing,Visualization;Fan Song,Shao-Yuan Liu,Xiao-Xiao Liu,Dan Mo,Yang Sun,Yuan-Yuan Gao,Wen Ye,Ying-Da Feng,Chang-Yang Xing,Chen Ye,andLei Zhou:Methodology,Investigation;Jing-Ru Meng:Methodology,Investigation,Supervision;Wei Cao:Funding acquisition,Conceptualization,Writing-Reviewing and Editing;Xiao-Qiang Li:Conceptualization,Resources,Funding acquisition,Conceptualization,Writing -Reviewing and Editing.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos.: 81970245,82270357,and 81770432),the Scientific Research Project of Shaanxi Administration of Traditional Chinese Medicine,China (Grant Nos.: 2021-04-ZZ-001,2021-QYPT-003,and 2022-SLRH-YQ-004),the Project of Science and Technology Department of Shaanxi Province in China(Project No.: 2022YWZX-PG-01),and the Natural Science Basic Research Program of Shaanxi Province in China (Grant No.: 2023-JC-JQ-61).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.08.018.
 Journal of Pharmaceutical Analysis2023年11期
Journal of Pharmaceutical Analysis2023年11期
- Journal of Pharmaceutical Analysis的其它文章
- Stage-specific treatment of colorectal cancer:A microRNA-nanocomposite approach
- Biosensors for waterborne virus detection: Challenges and strategies
- Development status of novel spectral imaging techniques and application to traditional Chinese medicine
- Oridonin restores hepatic lipid homeostasis in an LXRα-ATGL/EPT1 axis-dependent manner
- Ginsenoside Rg5 enhances the radiosensitivity of lung adenocarcinoma via reducing HSP90-CDC37 interaction and promoting client protein degradation
- Nanoscale coordination polymer Fe-DMY downregulatingPoldip2-Nox4-H2O2 pathway and alleviating diabetic retinopathy